Article Text

Abstract
Objective To evaluate the safety, tolerability, pharmacokinetics (PK) and preliminary antitumour activity of AMG 404, a fully human IgG1 monoclonal antibody targeting programmed cell death-1, in patients with advanced solid tumours.
Design First-in-human phase I study comprising eight dose expansion cohorts, including cohorts with microsatellite instability-high (MSI-H) tumours and non-small cell lung cancer with high programmed death-ligand 1 expression (NSCLC/PDL1-H, tumour proportion score ≥50%).
Setting Conducted across 28 global sites.
Participants This study enrolled adult patients with histologically or cytologically confirmed metastatic or locally advanced solid tumours not amenable to curative treatment with surgery or radiation. The inclusion criteria included a life expectancy of >3 months, ≥1 measurable or evaluable lesion per modified Response Evaluation Criteria in Solid Tumours (RECIST) V.1.1, an Eastern Cooperative Oncology Group performance status of ≤2 and adequate haematological, renal and hepatic function. Patients with prior treatment with checkpoint inhibitors, primary brain tumour or untreated or symptomatic brain metastases and leptomeningeal disease and history of other malignancy within the past 2 years were excluded.
Interventions The planned doses were 240 mg, 480 mg and 1050 mg of AMG 404 administered every 4 weeks (Q4W).
Primary and secondary outcome measures Primary endpoints were dose-limiting toxicities (DLTs), treatment-emergent adverse events, treatment-related adverse events, changes in vital signs and clinical laboratory tests. Secondary endpoints included PK parameters, incidence of antidrug (AMG 404) antibodies and antitumour activity assessed per modified RECIST V.1.1 (objective response, duration of response, progression-free survival (PFS), disease control and duration of stable disease).
Results A total of 171 patients were enrolled; 168 were treated. Median (range) follow-up was 36.3 weeks (1.6–137.1). No DLTs were observed. Grade 3 and serious treatment-related adverse events occurred in 16 (9.5%) and 12 (7.1%) patients, respectively. The 480 mg Q4W dose was selected as the recommended phase II dose. AMG 404 serum exposure increased approximately dose proportionally. The objective response rate (80% CI) was 19.6% (15.7–24.1) for the overall population and 36.6% (26.4–47.8) and 30.8% (14.2–52.3) for cohorts with MSI-H tumours (n=41) and NSCLC/PDL1-H (n=13), respectively. The overall disease control rate (80% CI) was 54.8% (49.5–59.9). The median (80% CI) PFS was 3.7 (3.5–4.5) months for the overall population and 14.8 (9.0–not estimable) and 4.4 (2.2–9.7) months for cohorts with MSI-H tumours and NSCLC/PDL1-H, respectively.
Conclusions AMG 404 monotherapy was tolerable at the tested doses, with encouraging antitumour activity observed across tumour types.
Trial registration number NCT03853109.
- Gastrointestinal tumours
- Molecular aspects
- Respiratory tract tumours
- IMMUNOLOGY
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
This study used a first-in-human dose-selection strategy using existing clinical experience of other programmed cell death-1 inhibitors to rapidly evaluate AMG 404 with a minimal study design of only three dose-escalation cohorts.
The trial prospectively enrolled patients, including those with prespecified histology and/or biomarkers (eg, dMMR/MSI-H tumours) that are particularly important to consider with immune checkpoint inhibitors.
This study had a relatively large enrolment for a phase I trial; however, individual cohorts were small to moderate in size, which may limit data interpretation.
This single-arm, open-label study was not powered to compare tumour types.
Fresh and archival biopsies were accepted, so biomarker profiles from some biopsies were not time-matched to the start of the trial.
Background
The programmed cell death-1 (PD-1) receptor-ligand interaction is a major pathway used by tumours to suppress immune control.1 PD-1 and cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) are co-inhibitory receptors expressed on T cells to negatively regulate antigen receptor signalling on engagement of its ligands (programmed death-ligand 1 (PD-L1) or 2).1–4 Overexpression of PD-L1 by cancer cells and PD-1 and CTLA-4 by T cells can be seen in the tumour microenvironment (TME).5 6 Tumour cells (TCs) exploit these immune checkpoint molecules to induce tumour tolerance and T-cell exhaustion.5 Immune checkpoint inhibitors (ICIs) can therefore restore effector T-cell function leading to antitumour activity.6
ICIs such as anti-PD-1 agents (eg, nivolumab, pembrolizumab, cemiplimab, sintilimab, toripalimab and tislelizumab), anti-PD-L1 agents (eg, atezolizumab, avelumab and durvalumab) and CTLA-4 inhibitors (eg, ipilimumab and tremelimumab) have demonstrated clinical efficacy in several advanced cancers.7–12 Furthermore, the efficacy of PD-1 blockade was demonstrated in cancers with DNA mismatch repair-deficient (dMMR) and microsatellite instability-high (MSI-H) status, a distinct subset with variable outcomes on standard therapies.13–15 Despite these promising outcomes, low and variable response rates with ICIs remain a clinical challenge, with 20%–40% of patients overall deriving clinical benefit.16
AMG 404 is a human IgG1 monoclonal antibody (mAb) with high affinity, potency and binding ability for PD-1, inhibiting receptor-ligand interaction. The fragment crystallisable (Fc) region was modified to eliminate undesired interactions with Fcγ receptors and complement. In cell-based assays, AMG 404 did not show residual antibody-dependent cellular cytotoxicity.17
We sought to design a study that would rapidly evaluate the clinical dosing for AMG 404 by leveraging clinical results for other anti-PD-1 mAbs at their approved doses from the literature and preclinical AMG 404 data to guide first-in-human (FIH) dose selection. This phase I trial (NCT03853109) evaluated the safety, pharmacokinetics (PK) and antitumour activity of AMG 404 in patients with advanced solid tumours. Herein, we report the primary analysis of the study.
Methods
Design and setting
This was an FIH, open-label, nonrandomised, multicentre, dose exploration and expansion, phase I study of AMG 404 in patients with advanced solid tumours conducted across 28 global sites (online supplemental figure 1 and online supplemental table 1). The study comprised nine dose exploration and expansion cohorts. As per the study protocol, Cohort 5, which included recommended phase II dose (RP2D) expansion in China/Taiwan/Hong Kong, was excluded from this analysis. The remaining cohorts were renumbered for clarity herein (online supplemental figure 1); Cohorts 5–8 detailed in the manuscript refer to Cohorts 6–9 in the protocol and ClinicalTrials.gov (online supplemental table 2). Further details are provided in the protocol (online only) and online supplemental file 1.
Supplemental material
The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. Institutional review boards or independent ethics committees at each participating site approved the protocol and its amendments (online supplemental table 1).
Patients
All enrolled patients provided written informed consent. This study enrolled patients (aged ≥18 years) with histologically or cytologically confirmed metastatic or locally advanced solid tumours not amenable to curative treatment with surgery or radiation. The inclusion criteria were as follows: a life expectancy of >3 months in the opinion of the investigator; ≥1 measurable or evaluable lesion per modified Response Evaluation Criteria in Solid Tumours (RECIST) V.1.1,18 which had not undergone biopsy within 3 months of the screening scan; an Eastern Cooperative Oncology Group performance status of ≤2; adequate haematological, renal and hepatic function; a negative pregnancy test for females of childbearing potential. Additional cohort-specific inclusion criteria included having one of the tumour types as specified in the study schema (Cohort 6), having MSI-H or dMMR tumours (Cohort 7) and having PD-L1-positive non-small cell lung cancer (NSCLC) (tumour proportion score (TPS) ≥50%) without EGFR or ALK or ROS1 genomic tumour aberrations and without receipt of prior systemic treatment for advanced disease (prior neoadjuvant, adjuvant or concurrent chemoradiation was permitted; Cohort 8). The specific tumour types in Cohort 6 included melanoma; small cell lung cancer; NSCLC; head and neck squamous cell carcinoma; urothelial, gastric or gastro-oesophageal junction adenocarcinoma; oesophageal, cervical or hepatocellular carcinoma; Merkel cell carcinoma; squamous cell carcinoma of the skin; renal cell carcinoma; sarcoma; thymic carcinoma; nasopharyngeal carcinoma; mesothelioma.
Key exclusion criteria included prior treatment with ICIs; having a primary brain tumour or untreated or symptomatic brain metastases and leptomeningeal disease; history of other malignancy within the past 2 years, with prespecified exceptions; history of solid organ transplantation; major surgery within 28 days of study day 1; antitumour therapy within 21 days before study day 1. Full details on eligibility criteria are provided in the supplement (online supplemental methods and study oversight, section on “Patients”).
Treatment and dosage
AMG 404 was administered via intravenous infusion every 4 weeks (Q4W). The following doses were planned: Cohort 1, 240 mg; Cohort 2, 480 mg; Cohort 3, expansion at 480 mg; Cohort 4, exploratory dose of 1050 mg; Cohort 5, expansion at the RP2D; Cohorts 6–8, expansion at RP2D in patients with prespecified histology and/or biomarkers as described above (online supplemental figure 1).
Treatment was continued until disease progression (PD), treatment intolerance, consent withdrawal or up to 2 years. In the final protocol, and prior to the sponsor’s decision to close the trial early, patients with a complete response (CR), partial response (PR), stable disease or continued clinical benefit at 2 years of treatment had the option to continue treatment for up to 4 years. Treatment could be temporarily withheld or discontinued as necessary, but a partial dose reduction was not permitted.
FIH dose selection
Starting dose and predicted efficacious dose were determined based on exposures predicted to provide similar PD-1 inhibition pharmacological activity to other anti-PD-1 mAbs at their approved doses that are efficacious and generally well tolerated in patients. AMG 404 efficacious exposure targets were predicted using exposures of pembrolizumab and nivolumab at their approved doses and adjusting for potency using in vitro PD-1/PD-L1 blockade activity of these mAbs. An AMG 404 dose of 480 mg Q4W was predicted to achieve the efficacious exposure targets using human PK predictions based on allometric scaling of AMG 404 PK in cynomolgus monkeys. The prediction was supported by the evaluation of in vitro pharmacological activities of AMG 404 and nivolumab, which is approved for dosing at 480 mg Q4W. A starting dose of 240 mg Q4W was selected using the efficacious dose estimate of 480 mg Q4W and addition of a twofold safety factor given the lack of correlation between nonclinical findings and clinical toxicity with PD-1 inhibitors.
Dose-limiting toxicity evaluation and dose escalation
The dose-limiting toxicity (DLT) evaluation period was the first 28 days of AMG 404 treatment. Dose escalation decisions were guided primarily by observed safety and tolerability of AMG 404. Dose level decisions followed the modified toxicity probability interval (TPI) model with a target TPI of 0.20–0.33, with TPI >0.33 defined as excessive toxicity.19
Endpoints
Primary endpoints were the patient incidence of DLTs, treatment-emergent adverse events (AEs), treatment-related adverse events (TRAEs), changes in vital signs and clinical laboratory tests. Secondary endpoints included PK parameters of AMG 404 including, but not limited to, maximum observed serum concentration (Cmax), time to achieve Cmax (Tmax), area under the serum concentration-time curve; patient incidence of antidrug (AMG 404) antibodies (ADAs); objective response (assessed every 8 weeks); duration of response (DOR); progression-free survival (PFS); disease control rate; duration of stable disease measured by CT/MRI and assessed per modified RECIST V.1.1.
Exploratory endpoints included the assessment of PD-L1, MSI status, tumour mutational burden (TMB) and gene expression signatures of tumour inflammation score (TIS) and interferon gamma score (IFNγ) score.
Assessments
Safety assessments included patient incidence of AEs using the Common Terminology Criteria for Adverse Events V.5.0.
For PK assessments, blood samples were collected before the first dose and at multiple time points thereafter across the first two dosing intervals. PK parameters were estimated using noncompartmental methods. Serum AMG 404 concentrations were measured using a validated electrochemiluminescence (ECL) immunoassay (lower limit of quantification, 10 ng/mL).
For ADA assessments, blood samples were collected before the first dose and at multiple time points thereafter, at the end of the study and at safety follow-up. A validated ECL bridging immunoassay was used to detect and quantify the serum levels of ADAs.
Best overall responses (BORs) were assessed using modified RECIST V.1.1 and did not require confirmation of PD; confirmation of CR and PR was required. The study used a modification to RECIST V.1.1 criteria which allowed up to five target lesions per organ and 10 total to increase lesion sampling and reduce assessment error.18
For biomarker assessments, PD-L1 TC expression was determined using the Ventana SP263 immunohistochemistry assay. Both TIS and IFNγ signatures were generated using whole transcriptome sequencing through the Tempus next-generation sequencing (NGS) platform. MSI status was determined via central polymerase chain reaction (Promega) or NGS (Tempus xT panel) testing. The TMB score was also determined via NGS (Tempus xT panel) testing.
Statistical analysis
This study had a planned enrolment of up to 275 evaluable patients, ranging from approximately 2–40 patients in each cohort, to provide a prespecified probability of observing ≥1 DLT (if the true DLT rate is 20%–33%) or ≥1 AE with 5%–10% incidence rate, depending on the cohort. All endpoints, unless noted otherwise, were assessed using the safety analysis set, defined as all enrolled patients who received ≥1 dose of AMG 404.
Descriptive statistics were provided for demographics, safety, PK parameters and biomarkers by dose and time, as appropriate.
Data summaries and efficacy and safety analyses were planned separately for prespecified cohorts versus other cohorts, with futility analysis planned for predetermined cohorts. The Clopper-Pearson CIs for proportions were estimated. For time-to-event endpoints, Kaplan-Meier estimates of the median and percentiles were assessed, with CIs calculated using the Brookmeyer and Crowley method. The biomarker analysis set included patients with biomarker data and an evaluable BOR (eg, PD, SD, PR and CR) evaluated using modified RECIST V.1.1. The Wilcoxon rank sum test was used for continuous biomarker data, and the Fisher exact test was used for binary biomarker data (eg, MSI and PD-L1) comparison between the responder and non-responder groups.
Patient and public involvement statement
Patients and/or the public were not involved in the design, conduct, reporting or dissemination plans of this research.
Results
Patients
Between March 2019 and January 2022, a total of 225 patients were screened and 171 were enrolled, of whom 168 were treated. Study enrolment was stopped early due to sponsor decision and not related to safety or lack of clinical efficacy. As of 19 July 2022 data cut-off, all patients who had received ≥1 dose of AMG 404 were included in the safety analysis set. About half (53.0%) of the overall patient population (all dose cohorts, n=168) were male, and the median (range) age was 62 years (26‒84) (table 1). The solid tumour types included gastrointestinal tumours in 59 (35.1%) patients, thoracic and head and neck tumours in 37 (22.0%), gynaecological tumours in 25 (14.9%), sarcomas in 23 (13.7%), other types of solid tumours in 18 (10.7%) and tumours with renal histology in 6 (3.6%). Overall, 142 (84.5%) patients had received prior anticancer therapy (53 (31.5%), 34 (20.2%), 25 (14.9%) and 30 (17.9%) patients received 1, 2, 3 and >3 lines of therapy, respectively; table 1). The median (range) number of lines of prior therapy in any setting was 2 (1–7). At the data cut-off, 5 (2.9%) patients completed the study treatment, and 132 (77.2%) discontinued treatment. PD (97/171 (56.7%)) was the most common reason for treatment discontinuation. The median (range) follow-up was 36.3 weeks (1.6–137.1).
Baseline demographics and characteristics
DLTs and safety
At the data cut-off, patients had received a median (range) cumulative dose of 2400 mg (480–23,100) AMG 404, with a median (range) treatment duration of 15.6 weeks (0.1–133.4). A median (range) of 4.5 cycles (1–31) of AMG 404 were initiated. DLTs were evaluated in Cohorts 1–4 to determine the RP2D. In these cohorts, no DLTs were reported during the DLT evaluation period.
AEs, regardless of attribution, occurred in all patients who received AMG 404 except one patient (online supplemental table 3). (7.1%) patients discontinued treatment due to treatment-emergent AEs, of whom 10 (6.9%) were in the 480 mg cohort and 2 (9.5%) in the 1050 mg cohort. Overall, 109 (64.9%) patients experienced ≥1 TRAE of any grade; the most common ones were fatigue (16.1%) and pruritus (10.7%; table 2). Grade 3 and serious TRAEs occurred in 16 (9.5%) and 12 (7.1%) patients, respectively; 5 (3.0%) patients discontinued treatment due to TRAEs, all of whom were in the 480 mg cohort. No Grade 4 TRAEs were reported. A fatal TRAE due to metastasis to the spine was reported in 1 (0.7%) patient in the 480 mg cohort.
Summary of TRAEs
PK
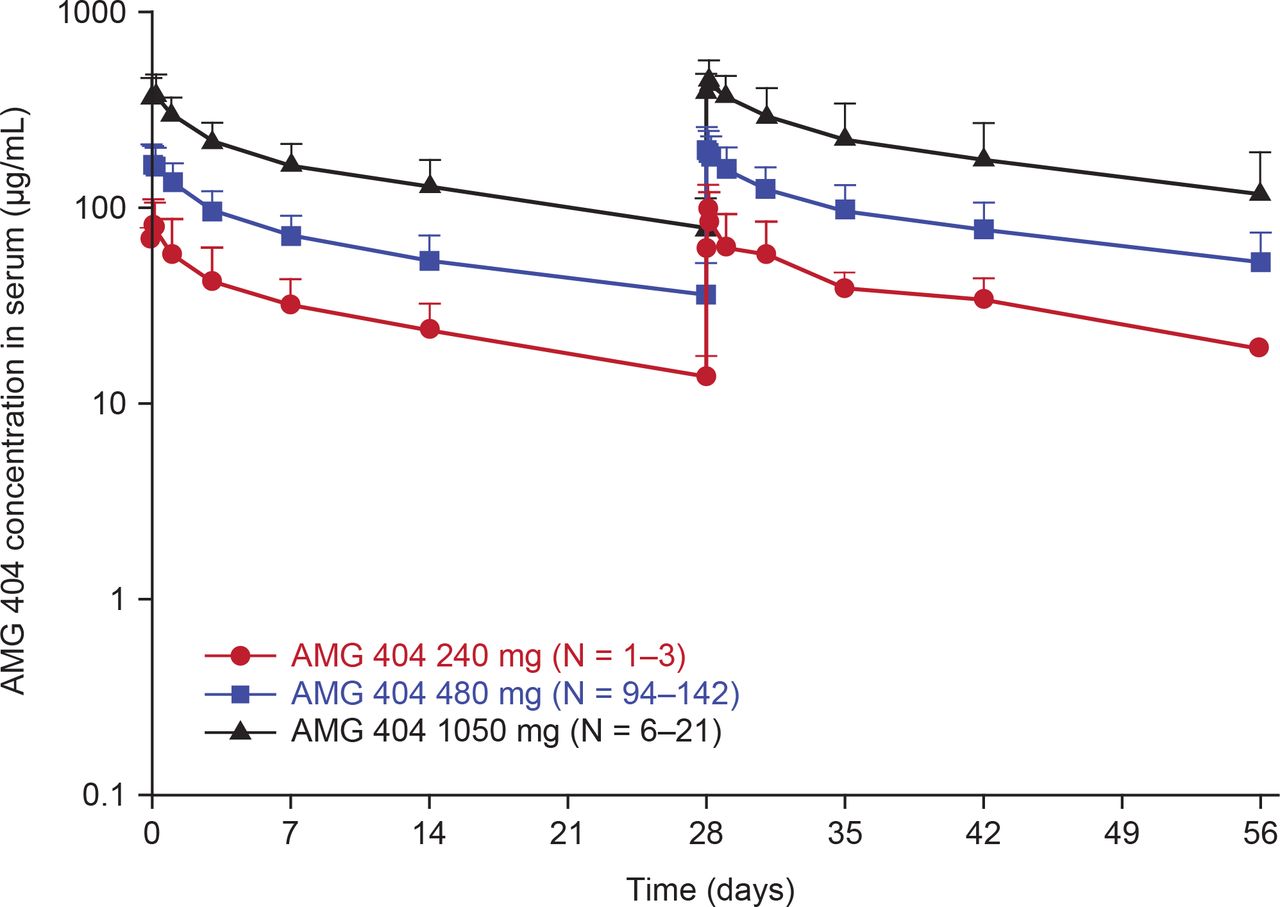
Samples for PK analysis were collected from a total of 168 patients at the planned time points. An approximately dose-proportional increase in exposure was observed in the dose range of 240–1050 mg Q4W (figure 1). Minimal serum accumulation of AMG 404 was observed with repeat Q4W dosing.

Mean (±SD) serum concentration-time profiles of AMG 404 following once every 4 week intravenous administration of AMG 404 for the first two dosing intervals. IV, intravenous; Q4W, every 4 weeks.
Immunogenicity
A total of 158 patients had postbaseline samples and were therefore evaluable for ADA assessments. Of these patients, 27 (17.1%) were positive for treatment-emergent AMG 404 ADAs. Of the 27 ADA-positive patients, one was in the 1050 mg dose cohort, while the rest were in the 480 mg dose cohort; 19 (70.4%) patients had an ADA response that was transient. A total of 166 patients had ADA results at baseline, of whom 3 (1.8%) had pre-existing AMG 404 ADAs, none of which were boosted on AMG 404 treatment.
Clinical activity
In the overall population, the ORR (80% CI) was 19.6% (15.7–24.1), with 2.4% and 17.3% of patients achieving CR and PR, respectively. ORRs (80% CI) in the 240 mg, 480 mg and 1050 mg cohorts (n=3/144/21) were 0%, 22.2% (17.8–27.3) and 4.8% (0.5–17.3), respectively, with a CR in 4 (2.8%) patients in the 480 mg cohort only (table 3). When assessed by tumour type, the ORR (80% CI) was 9.6% (5.4–15.6), 17.1% (9.8–27.0), 36.6% (26.4–47.8) and 30.8% (14.2–52.3) for Cohorts 1–5 (any tumour type; n=73; 2 CR, 5 PR), Cohort 6 (selected tumour types; n=41; 7 PR), Cohort 7 (MSI-H tumours; n=41; 2 CR, 13 PR) and Cohort 8 (NSCLC/PDL1-H; n=13; 4 PR), respectively (online supplemental table 4).
Tumour response to AMG 404 per modified RECIST V.1.1 in solid tumours by dose levels
The DCR (80% CI) was 54.8% (49.5–59.9) for all cohorts combined and 66.7% (19.6–96.6), 56.3% (50.6–61.8) and 42.9% (27.8–59.1) for patients in the 240 mg, 480 mg and 1050 mg cohorts, respectively (table 3). The DCR (80% CI) was 45.2% (37.2–53.4), 48.8% (37.8–59.9), 70.7% (59.8–80.1) and 76.9% (55.6–91.2) for Cohorts 1–5 (any tumour type), Cohort 6 (selected tumour types), Cohort 7 (MSI-H tumours) and Cohort 8 (NSCLC/PDL1-H), respectively (online supplemental table 4). The median (80% CI) DOR in responders was 16.9 months (not estimable (NE)) in the 1050 mg cohort and was not reached in the 480 mg cohort (data not shown). Patients in the 240 mg cohort did not achieve an objective response and were therefore not considered for DOR assessment.
The median (80% CI) PFS per modified RECIST v1.1 was 3.7 months (3.5–4.5), with an estimated 12-month PFS rate of 26.7% (80% CI: 22.2–31.5) for all cohorts combined. The median (80% CI) PFS was 2.9 (1.5–NE), 3.7 (3.5–5.5) and 1.8 (1.7–4.5) months in the 240 mg, 480 mg and 1050 mg cohorts, respectively (table 3). The median (80% CI) PFS was 2.8 (1.9–3.7), 3.0 (1.9–4.3), 14.8 (9.0–NE) and 4.4 (2.2–9.7) months for Cohorts 1–5 (any tumour type), Cohort 6 (selected immunosensitive tumour types), Cohort 7 (MSI-H tumours) and Cohort 8 (NSCLC/PDL1-H), respectively (online supplemental table 4).
Biomarker and clinical outcome
At least one central biomarker result was obtained from 144 patients; biomarker ascertainment by cohort is described in online supplemental table 5. Patients with tumour tissue expression of PD-L1 TC ≥1% had greater ORR and a superior median (80% CI) PFS of 7.2 (4.9–12.9) months compared with that of 1.9 (1.9–3.5) months in patients with TC <1% (figure 2). Patients with MSI-H tumours had greater ORR and a longer median (80% CI) PFS of 14.8 (9.3–20.5) months compared with that of 2.4 (1.9–3.7) months in patients with microsatellite stable tumours (figure 2). Higher TIS and IFNγ scores, indicative of a more inflamed TME, were associated with improved tumour response (online supplemental figure 2). Additionally, higher median TMB levels were associated with clinical response (online supplemental figure 2).

{kind=link}
{kind=link}
Association between biomarkers PD-L1 and MSI with clinical response (CR), complete response, microsatellite instability (MSI), MSI-high (MSI-H), microsatellite stability (MSS), nonresponders (NR), progressive disease (PD), programmed death-ligand 1 (PD-L1), progression-free survival (PFS), partial response (PR), stable disease (SD) and tumour cell (TC).
Discussion
In this FIH phase I study of the PD-1 inhibitor AMG 404 in patients with advanced solid tumours, AMG 404 was well tolerated at the doses tested; the 480 mg Q4W dose was determined to be the RP2D. No DLTs were reported. The majority of AMG 404-related AEs observed were Grade 1–2. Clinical responses were observed across multiple cancer types, including dMMR tumours and NSCLC/PDL1-H. The PK results were consistent with those of prior studies of therapeutic anti-PD-1 mAbs.7 8 11 20–22 A positive association of several tumour biomarkers with clinical response was observed.
The safety profile was consistent with that observed for other PD-1 inhibitors, with commonly reported AEs of fatigue, nausea, decreased appetite and constipation.7 8 11 21 TRAEs occurred in 64.9% of patients, which is generally comparable to the incidence rates with pembrolizumab (70.0%–80.0%), sintilimab (54.3%), toripalimab (88.0%) and tislelizumab (57.4%) in advanced solid tumours.7 11 12 23 24 Differences in incidences or severities of AEs were observed among the three dose groups, although the more limited sample sizes of the 240 mg and 1050 mg cohorts limit data interpretation. The overall treatment discontinuation rate due to TRAEs was low (3.0%), indicating a manageable safety profile in this population.
A dose of 480 mg Q4W was selected as the RP2D based on the safety, PK and efficacy profiles between dose levels ranging from 240 mg to a maximum of 1050 mg Q4W. This approach enabled us to confirm the safety of a higher dose of AMG 404 and demonstrate that the efficacy of AMG 404 is unlikely to increase with the maximum dose. Indeed, no CRs were observed in the 1050 mg cohort, and the ORR and DCR were numerically higher in the 480 mg cohort than in the 1050 mg cohort. These findings are consistent with the flat exposure-efficacy relationships observed for pembrolizumab and nivolumab over a range of exposures resulting from doses at and above their approved doses.25 26 Further, population PK analyses indicate that body weight was identified as a covariate of AMG 404 PK (data not shown), consistent with other PD-1 inhibitors.27 However, the covariate effect for AMG 404 was within range for other mAbs where both fixed and body weight-based dosing approaches were shown to perform similarly, supporting no clinically significant effect of body weight on PK and the use of fixed dosing for AMG 404.28
The 17.1% incidence of ADAs with AMG 404 is numerically higher than that reported with nivolumab (12.7%), pembrolizumab (0.7%‒2.5%) and cemiplimab (1.3%–2.0%).8 29 However, the ADA responses were transient in the majority of patients, suggesting that they did not likely affect AMG 404 treatment outcomes. The overall ORR of 19.6% is generally similar (or higher) to that shown with other PD-1 inhibitors (cemiplimab, 10.0%–22.2%; sintilimab, 12.6%; toripalimab, 7.1%–12.5%; tislelizumab, 13.3%).7 8 10–12 30 The ORR observed in the 480 mg cohort was numerically higher than that observed in the 1050 mg cohort, consistent with no increase in clinical activity with increase from the 480 mg dose to the 1050 mg dose, although the 1050 mg cohort size was relatively limited (n=21). It is inconclusive whether the lack of objective responses in the 240 mg cohort was related to the dose level or was a result of the small cohort size and/or interpatient variability. In addition, cohorts with NSCLC/PDL1-H and MSI-H tumours (including mCRC, n=~20) appeared to derive particular benefit. The NSCLC/PDL1-H cohort demonstrated an ORR and DCR of 30.8% and 76.9%, respectively. This is generally comparable with the ORR reported with cemiplimab (25.0%) in patients with pretreated advanced NSCLC, although the Moreno et al study assessed ORR by independent central review and had a majority of patients with a PD-L1 TPS of <1% with only 16.7% having a ≥50% TPS.31 The dMMR/MSI-H tumour cohort showed an ORR and DCR of 36.6% and 70.7%, respectively, which are comparable to the treatment outcomes with nivolumab and pembrolizumab in dMMR mCRC and dMMR non-colorectal cancer patient subsets.13–15 24
The overall DCR of 54.8% with AMG 404 is generally comparable with those from other PD-1 reports (cemiplimab, 50.0%; sintilimab, 44.2%; toripalimab, 20.8%–39.3%; tislelizumab, 44.6%).7 10–12 31 The overall median PFS with AMG 404 was 3.7 months, which is numerically longer than that reported with other PD-1 inhibitors (toripalimab, 2.8 months; sintilimab, 1.6 months; tislelizumab, 2.1 months), with all studies having used RECIST V.1.1 for response evaluation similar to the current study.7 10 12 Furthermore, the 12-month PFS rate of 26.7% in this study is higher than that observed with sintilimab (10.4%) and tislelizumab (15.6%).7 12
Associations of AMG 404 clinical response with relevant biomarkers were also assessed. Tumours with high PD-L1 expression and TIS are indicative of a T cell-inflamed TME, while high TMB and MSI-H status reflect a hypermutated tumour phenotype that may lead to increased neoantigen presentation and immune response.32 33 The expression levels of these biomarkers have been described to predict ICI efficacy.32 34 35 PD-L1 is currently the most widely validated and adopted biomarker for immunotherapy outcomes.36 IFNγ signalling is a predictive correlate of response to anti-PD-1/PD-L1 therapy.37–39 A significant correlation between TMB and ORRs with anti-PD-1/PD-L1 monotherapy was shown across tumour types.40 The 18-gene TIS retrospectively predicted clinical benefit in pembrolizumab clinical trials.37 TMB and TIS were independently predictive of response to pembrolizumab using KEYNOTE trial datasets.41 In the current study, these biomarkers showed a differential expression in responders to AMG 404 compared with nonresponders. Responders were enriched with patients whose tumours had higher TMB score, TIS score, IFNγ-related gene signature, PD-L1 protein expression (TPS, ≥ 1 vs <1) or MSI-H status than nonresponders, consistent with previous ICI studies.37 41–43
Limitations of this study include the single-arm, open-label study design, which was not powered to compare tumour types. Given the relatively small cohort sizes and tumour-agnostic nature of the protocol, all biomarker-evaluable patients were grouped and analysed together without accounting for tumour type. Fresh and archival biopsies were accepted, so biomarker profiles from some biopsies were not time-matched to the start of the trial. Furthermore, the relatively small sample sizes of the 480 mg and 1050 mg cohorts limit the interpretation of data. Despite these limitations, this study has several strengths. First, the study design allowed for tumour-specific cohorts and biomarker-enriched cohorts. Second, preclinical PK modelling and comparison with other PD-1 inhibitors were used as a basis to begin the trial at one dose level below the predicted efficacious dose, limiting exposure of patients to doses that are not expected to be efficacious. Further, though PD-1 drugs are well-characterised, translation of in vitro PD-1/PD-L1 blockade activity of new and existing PD-1 inhibitors to predict the efficacious dose of the new PD-1 inhibitor in patients was unknown. The observed clinical efficacy with AMG 404 confirming the predicted dose of 480 mg Q4W as efficacious in patients supports this approach. Third, this study had a relatively large enrolment for a phase I trial, which further helped support that starting at a near-efficacious dose was accurate. Fourth, the trial prospectively enrolled patients with dMMR/MSI-H tumours. Finally, these efficacy and safety data for AMG 404 support the successful FIH dose selection strategy using existing clinical experience of other PD-1 inhibitors to rapidly evaluate AMG 404 with an efficient and minimal study design of only three dose escalation cohorts. The design included cohorts for (1) a starting dose one level below the RP2D for safety purposes given the lack of correlation between nonclinical and clinical toxicity for PD-1 inhibitors, (2) the RP2D and (3) one dose level above the RP2D to establish a safety margin. This strategy can be applied to new investigational drugs for other drug classes where existing clinical data in the same drug class are available.
Conclusions
AMG 404 monotherapy at doses of 240 mg, 480 mg and 1050 mg Q4W showed no DLTs and was generally well tolerated in patients with advanced solid tumours. Encouraging antitumour activity was observed. The observed AEs, PK profiles and biomarker trends of AMG 404 were consistent with prior studies of ICIs. These data support that patients with advanced solid tumours, including unique, biomarker-defined, clinically relevant subsets such as MSI-H status and NSCLC/PDL1-H, may benefit from PD-1 blockade immunotherapy with AMG 404.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by the institutional review boards or ethics committees of each participating site as listed in supplemental table S1 provided in the supplemental file available online and below. All patients provided written informed consent. Reference or approval numbers cannot be found in online supplemental table S1.
Acknowledgments
We thank the patients, investigators and study staff who contributed to this study. Medical writing support was provided by Shubha Dastidar, PhD, CMPP (Cactus Life Sciences, part of Cactus Communications), and was funded by Amgen Inc. The study was sponsored and funded by Amgen Inc.
References
Footnotes
Contributors Conception and design: TP, MK, HW and HP. Provision of study materials or patients: TP, IL, SPC, GF, VS, JGM, H-TA, MH, YK, RD, RS, MHH, DT, CML and HP. Collection and assembly of data: TP, IL, SPC, GF, VS, JGM, H-TA, MH, YK, RD, RS, MHH, DT, CML, MK, AH, WS and HP. Data analysis and interpretation: KW, AH, WS, HW and MK. Manuscript writing: all authors. Final approval of manuscript: all authors. Accountable for all aspects of the work: all authors. Guarantor is TP.
Funding This study was sponsored by Amgen Inc.
Disclaimer The funder was involved in the design of the trial; collection, analysis and interpretation of data; development of the manuscript.
Competing interests TP: honorarium: Amgen, Servier, MSD; advisory board: Amgen, Servier, MSD and BMS. IL: Roche, MSD, Amgen, BMS, Pfizer, Ryvu, Takeda Siropa, Rhizen, Menarini, Incyte and MacroGenics; grants or contracts: Roche, Agenus and ABM; payment or honoraria for lectures, presentations, speaker bureaus, manuscript writing or educational events: Roche and MSD; support for attending meetings and/or travel: Roche; participation on a data safety monitoring board or advisory board: Roche, MSD, Amgen, BMS, Pfizer, Ryvu, Takeda Siropa, Rhizen, Menarini, Incyte and MacroGenics; leadership or fiduciary role in other board, society, committee or advocacy groups, paid or unpaid: ESMO, OECI, EURACAN and EUMelaReg; other financial or non-financial interests: CliniNote. SPC: grants or contracts: Amgen, Roche, GSK, Threshold Pharmaceuticals, CytRx Corporation, Ignyta, Immune Design, TRACON Pharma, Karyopharm Therapeutics, Sarcoma Alliance for Research through Collaboration (SARC), Janssen, Advenchen Laboratories, Bayer, Inhibrx, NKMax and Thyme; consulting fees: Amgen, Roche, GSK, Threshold Pharmaceuticals, CytRx Corporation, Ignyta, Immune Design, TRACON Pharma, Karyopharm Therapeutics, Sarcoma Alliance for Research through Collaboration (SARC), Janssen, Advenchen Laboratories, Bayer, Inhibrx, NKMax and Thyme; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events: Amgen, Roche, GSK, Threshold Pharmaceuticals, CytRx Corporation, Ignyta, Immune Design, TRACON Pharma, Karyopharm Therapeutics, Sarcoma Alliance for Research through Collaboration (SARC), Janssen, Advenchen Laboratories, Bayer, Inhibrx, NKMax and Thyme; stock or stock options: AADi, Cellestia Biotech, CounterPoint and Immix Biopharma. GF: royalties (self): Wolters Kluwer (2014–present); advisory role (to institution): AbbVie (2022), Fujifilm (2018), Silicon (2020, 2021), Navire (2021), Turning Point (2021), Predicine (2021), Inspirna (2021), Regeneron (2021), Jubilant (2022), BostonGene (2022), Teon (2022), Merck (2022), Sanofi (2023) and BridgeBio (2023); advisory role (self): EMD Serono (2010, 2011); speakers honorarium for CME: Total Health Conferencing (2019), Rocky Mountain Oncology Society (2020); travel (self, for work and/or research related to institution): Amgen (2022), Bristol Myers Squibb (2015), EMD Serono (2011, 2012, 2013), Fujifilm (2018), Millennium (2013), Sarah Cannon Research Institute (employer, at least once yearly) and Synthorx/Sanofi (2022); research funding [to institution, for any trial for which the author has been the principal investigator (ever) or sub-investigator (minimum last 4 years)]: 3-V Biosciences, Abbisko, Abbvie, ABL Bio, ADC Therapeutics, Accutar, Agenus, Aileron, American Society of Clinical Oncology, Amgen, ARMO/Eli Lilly, Artios, AstraZeneca, Bayer, BeiGene, BioAtla, BioInvent, Bio-Thera, Bicycle, Black Diamond, Boehringer Ingelheim, Celgene, Celldex, Ciclomed, Curegenix, Curis, Cyteir, Daiichi, DelMar, eFFECTOR, Eli Lilly, EMD Serono, Epizyme, Erasca, Exelixis, Freenome, Fujifilm, Genmab, GlaxoSmithKline, Hutchison MediPharma, IGM Biosciences, Ignyta, Immunitas, ImmunoGen/MacroGenics, Incyte, Jacobio, Jazz, Jounce, Jubilant, Kineta, Kolltan, Loxo/Bayer, MedImmune, Merck, Metabomed, Millennium, Mirati, miRNA Therapeutics, Molecular Templates, National Institutes of Health, Navire/BridgeBio, NGM Bio, NiKang, Novartis, OncoMed, Oncorus, Oncothyreon, Poseida, Precision Oncology, Prelude, PureTech, Pyramid, Pyxis, RasCal, Regeneron, Relay, Rgenix, Ribon, Roche, Samumed, Sapience, Seagen, Silicon, Simcha, Sirnaomics, Strategia, Syndax, Synthorx/Sanofi, Taiho, Takeda, Tallac, Tarus, Tarveda, Teneobio, Tesaro, Tocagen, Turning Point, U.T. MD Anderson Cancer Center, Vegenics, Xencor and Zhuhai Yufan. VS: research funding: Amgen. JGM: Research funding: Merck; advisory: BMS, Incyte, Merck, Pfizer, Sanofi, Taiho. H-TA: grants or contracts: Sarah Cannon Research Institute; payment or honoraria for lectures, presentations, speaker bureaus, manuscript writing or educational events: Servier; participation on a data safety monitoring board or advisory board: Labgenius, iOnctura and Engitix; stock or stock options: Ellipses Pharma. MH: research funding and honoraria: Roche; research funding: Pfizer; honoraria: MSD. YK: honoraria: Bristol Myers Squibb Japan, Lilly Japan and Taiho Pharmaceutical; consulting or advisory role: Amgen, Boehringer Ingelheim and Takeda; Research funding: Abbvie (Inst), Amgen (Inst), Astellas Pharma (Inst), AstraZeneca (Inst), Boehringer Ingelheim (Inst), Chugai Pharma (Inst), Genmab (Inst), GlaxoSmithKline (Inst), Incyte (Inst), Janssen Oncology (Inst), Lilly (Inst), Merck Serono (Inst), Ono Pharmaceutical (Inst), Taiho Pharmaceutical (Inst), Takeda (Inst), Hengrui (Inst) and Novartis Pharma (Inst): travel, accommodations and expenses: Amgen. RD: Amgen, Roche, AstraZeneca, Novartis, Takeda, MSD, Bristol Myers Squibb, GlaxoSmithKline, Janssen Pharmaceuticals and Boehringer Ingelheim. MHH: honoraria: Amgen, AstraZeneca, BMS, Janssen, MSD, Takeda and Roche; consulting or advisory role: AstraZeneca, BMS, MSD, Takeda, Roche and Yuhan; investigator or co-investigator of trials: Abbvie, Amgen, AstraZeneca, BMS, GSK, Ignyta, Janssen, Loxo Oncology, Merck Sereno, MSD, Novartis, Roche, Pfizer and Yuhan; research support: MSD, AstraZeneca and Yuhan. DT: grants and personal fees: Novartis, Bayer, AstraZeneca and Pfizer; personal fees: Boehringer Ingelheim, Eli Lilly, Loxo, Merck, Roche, Takeda and Merrimack; grants: GlaxoSmithKline, outside the submitted work. CML: Research funding: Amgen (Inst). KW, AH, WS, HW, MK: Employee and stockholder of Amgen Inc. HP: honoraria: Amgen, Roche, Sanofi, AstraZeneca and Bayer; consulting or advisory role: Biocartis and Cureteq.
Patient and public involvement Patients and/or the public were not involved in the design, conduct, reporting or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.