Article Text

Abstract
Introduction Postoperative acute kidney injury (AKI) is a common complication in cardiac surgery. Levels of intravascular haemolysis are strongly associated with postoperative AKI and with prolonged (>90 min) use of cardiopulmonary bypass (CPB). Ferrous plasma haemoglobin released into the circulation acts as a scavenger of nitric oxide (NO) produced by endothelial cells. Consequently, the vascular bioavailability of NO is reduced, leading to vasoconstriction and impaired renal function. In patients with cardiovascular risk factors, the endothelium is dysfunctional and cannot replenish the NO deficit. A previous clinical study in young cardiac surgical patients with rheumatic fever, without evidence of endothelial dysfunction, showed that supplementation of NO gas decreases AKI by converting ferrous plasma haemoglobin to ferric methaemoglobin, thus preserving vascular NO. In this current trial, we hypothesised that 24 hours administration of NO gas will reduce AKI following CPB in patients with endothelial dysfunction.
Methods This is a single-centre, randomised (1:1) controlled, parallel-arm superiority trial that includes patients with endothelial dysfunction, stable kidney function and who are undergoing cardiac surgery procedures with an expected CPB duration >90 min. After randomisation, 80 parts per million (ppm) NO (intervention group) or 80 ppm nitrogen (N2, control group) are added to the gas mixture. Test gases (N2 or NO) are delivered during CPB and for 24 hours after surgery. The primary study outcome is the occurrence of AKI among study groups. Key secondary outcomes include AKI severity, occurrence of renal replacement therapy, major adverse kidney events at 6 weeks after surgery and mortality. We are recruiting 250 patients, allowing detection of a 35% AKI relative risk reduction, assuming a two-sided error of 0.05.
Ethics and dissemination The Partners Human Research Committee approved this trial. Recruitment began in February 2017. Dissemination plans include presentations at scientific conferences, scientific publications and advertising flyers and posters at Massachusetts General Hospital.
Trial registration number NCT02836899.
- nitric oxide
- hemolysis
- acute kidney injury
- cardiopulmonary bypass
- endothelial dysfunction
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
Supplementation with nitric oxide (NO) gas during and after surgery requiring prolonged cardiopulmonary bypass might be most beneficial to those patients with endothelial dysfunction who are unable to replace the haemolysis-mediated NO depletion due to an impaired endothelial nitric oxide synthase (eNOS) activity.
The number of confounding variables resulting from the liberal use of nephrotoxic agents, the preoperative variability in serum creatinine level and the perioperative optimisation of haemodynamics and volume status is minimised, since the "Kidney Disease: Improving Global Outcomes"guidelines for renal protection are a standard of care at our institution.
Results from this study could possibly be generalised to other clinical scenarios of intravascular haemolysis and vascular depletion of NO.
This trial is designed to recruit patients from a single academic institution (Massachusetts General Hospital).
Introduction
Acute kidney injury (AKI) is a common and serious complication after cardiac surgery procedures requiring prolonged (>90 min) cardiopulmonary bypass (CPB).1–3 Because postcardiac surgical AKI is associated with increased risk of chronic kidney disease (CKD) and mortality,4–6 multiple interventions have been tested in large multicentre trials, but none have been successful at reducing AKI.7–10
There are two major categories of risk factors for postsurgical AKI. The first is the patient’s cardiovascular comorbidities that are associated with impaired endothelial function. These include obesity, diabetes, atherosclerosis, peripheral vascular disease, hyperlipidaemia, smoking and hypertension.2 11 The second is the extent of CPB-associated haemolysis.12–15 During haemolysis, plasma haemoglobin (Hb) is released into the circulation and depletes plasma nitric oxide (NO).16 17 NO is a potent mediator of vasodilation and regulates blood flow to tissues.18 In patients with endothelial dysfunction, the activity of endothelial NO synthase (eNOS), the enzyme responsible for NO production, is impaired and is unable to replenish the NO consumed by plasma Hb.19–21
In an animal model of free water-induced intravascular haemolysis, Minneci et al showed that haemolysis resulted in a reduction of creatinine clearance. The negative effects of haemolysis on renal function were attenuated by the inhalation of 80 parts per million (ppm) NO gas. NO oxidised plasma Oxy-Hb (Fe2+) to ferric methaemoglobin (Met-Hb, Fe3+), thereby decreasing plasma NO consumption.16 In a meta-analysis, the administration of NO gas has been associated with increased risk of AKI in critically ill patients with acute respiratory distress syndrome (ARDS), especially with prolonged use.22 However, no studies have shown an association between NO administration and increased incidence of AKI in paediatric or adult cardiac surgery population.
In a recent randomised controlled trial (RCT), administration of 80 ppm NO gas for 24 hours, starting at the onset of CPB, was found to decrease postoperative AKI in a population of young Chinese cardiac surgical patients (average age 48), with no risk factors for endothelial dysfunction, who were undergoing cardiac valve replacement due to rheumatic heart disease.17 NO administration also improved long-term outcomes such as major adverse kidney events (MAKE) and reduced stage 3 CKD for up to 1 year after surgery. However, it is still undetermined whether NO gas exerts similar, or even additional, renal protective effects in patients with endothelial dysfunction commonly treated in the USA.
Methods and analysis
Trial design
In this, single-centre, randomised (1:1) controlled, parallel-arm superiority trial, we hypothesised that 24 hours administration of NO gas starting from the initiation of CPB will decrease AKI by converting Oxy-Hb to Met-Hb in patients with signs and symptoms of endothelial dysfunction suggestive of impaired activity of eNOS (primary outcome).
After screening, consented patients who meet the enrolment criteria are randomised to receive either 80 ppm NO (intervention group, n=125) or nitrogen (N2, control group, n=125) for 24 hours as N2 is the major component of air and can be used as placebo. The test gas is administered via the oxygenator during CPB, then by inhalation once mechanical ventilation is resumed. When the patient is extubated, the test gas is delivered by high flow nasal cannula. On the day of surgery, after the insertion of a pulmonary artery catheter (PAC), patients are stratified by preoperative mean pulmonary artery pressure (mPAP) (ie, mPAP <30 mm Hg or mPAP ≥30 mm Hg) then allocated into the two study groups. Consented patients, with mPAP found to be ≥40 mm Hg on the day of surgery after PAC placement, are not randomised in this study because NO or other pulmonary vasodilators might be indicated as part of the medical treatment during the perioperative period. Study outcomes are assessed during the hospital stay, at a 6-week follow-up visit and by phone at 90 days and 1 year after the surgical procedure.
In order to avoid postoperative AKI, at our institution, the ‘Kidney Disease: Improving Global Outcomes (KDIGO) guidelines’ are the standard of care in all cardiac surgical patients.23 The implementation of these guidelines has been shown to limit the incidence and severity of CPB-associated AKI.24 Per these guidelines, patients do not receive any nephrotoxic agents such as intravenous contrast agents25 during the first 48 hours after surgery. Patients receive close monitoring of serum creatinine and hourly urine output and avoid intraoperative and postoperative hyperglycaemia for at least 72 hours after surgery. Haemodynamics are closely monitored to maintain adequate cardiac output (CO) and blood pressure by using intraoperative transoesophageal echocardiography (TEE) in combination with the PAC and a radial artery catheter.23 24 The pulmonary and radial artery catheters are removed only after the patient is discharged from the intensive care unit (ICU) and after completion of test gas treatment.
The study enrolment opened in February 2017 and the first patient entered the trial in June 2017. The study team plans to enrol 250 patients over a period of 3 years. The study design is depicted in figure 1.

Study design. After placement of pulmonary artery catheter, to ensure balance between study groups with respect to the likelihood of receiving NO after surgery, patients are randomised based on mPAP measured by the pulmonary artery catheter placed on the day of surgery (mPAP <30 mm Hg and mPAP between 30 mm Hg and 39 mm Hg). CPB, cardiopulmonary bypass; mPAP, mean pulmonary artery pressure; N2, nitrogen; NO, nitric oxide.
Eligibility criteria
The study population includes men aged >40 years and women aged >50 years who have signs and symptoms of endothelial dysfunction, have stable kidney function in the 3 months prior to surgery, are undergoing cardiac surgery procedures with expected prolonged CPB (>90 min) and are not currently enrolled in other research studies. The presence of endothelial dysfunction is evaluated by using a brief questionnaire that integrates medical history with clinical and laboratory data that have previously been associated with endothelial dysfunction (figure 2).26–35 Eligible patients are those undergoing a scheduled procedure or patients who are already admitted to the hospital. Key exclusion criteria include estimated glomerular filtration rate (eGFR) <30 mL/min/1.73 m2, mPAP ≥40 mm Hg and intravenous contrast infusion within 48 hours before surgery. A complete list of enrolment criteria is provided in box 1.

Screening questionnaire to detect endothelial dysfunction. The questionnaire above aims to systematically detect endothelial dysfunction in patients undergoing a cardiac surgical procedure. If ‘yes’ is answered to at least one of the above questions, the patient can be considered to have endothelial dysfunction and he/she may be enrolled in the study. BMI, body mass index; CABG, coronary artery bypass surgery; IDDM, insulin-dependent diabetes mellitus; LDL, low-density lipoprotein; NIDDM, non-insulin dependent diabetes mellitus; PTCA, percutaneous transluminal coronary angioplasty; SBP, systolic blood pressure.
Inclusion and exclusion criteria
Inclusion criteria:
Age ≥18 years.
Elective cardiac or aortic surgery requiring CPB and with expected CPB >90 min.
Clinical evidence of endothelial dysfunction assessed by a specifically designed questionnaire (figure 2).
Stable preoperative renal function without evidence of a plasma creatinine increase of ≥0.3 mg/dL within 3 months of study entry and without receiving RRT.
Exclusion criteria:
eGFR less than 30 mL/min/1.73 m2.
Emergent cardiac surgery.
Life expectancy <1 year at the time of enrolment.
mPAP ≥40 mm Hg and PVR >4 Wood Units.
LVEF <30% by echocardiography obtained within 3 months of enrolment.
Haemodynamic instability on the day of surgery as defined by a SBP <90 mm Hg.
Administration of one or more pRBC transfusion in the week prior to enrolment.
X-ray contrast infusion less than 48 hours before surgery.
Evidence of intravascular or extravascular haemolysis from any other origin:
Intravascular: intrinsic RBC defects leading to haemolytic anaemia (eg, enzyme deficiencies, haemoglobinopathies, membrane defects). Extrinsic: liver disease, hypersplenism, infections (eg, bartonella, babesia, malaria), treatment with oxidising exogenous agents (eg, dapsone, nitrites, aniline dyes), exposure to other haemolytic agents (eg, lead, snake and spider bites), lymphocyte leukaemia, autoimmune haemolytic disorders.
Extravascular: infection (eg, clostridial sepsis, severe malaria), paroxysmal cold hemoglobinuria, cold agglutinin disease, paroxysmal nocturnal hemoglobinuria, intravenous infusion of Rho (D) immune globulin, intravenous infusion of hypotonic solutions.
CPB, cardiopulmonary Bypass; eGFR, estimated glomerular filtration rate67; LVEF, left ventricular ejection fraction; mPAP, mean pulmonary artery pressure; pRBC, packed red blood cells; PVR, pulmonary vascular resistance; RRT, renal replacement therapy; SBP, systolic blood pressure.
Intervention
Perioperative management
Perioperative management including the implementation of the KDIGO guidelines has been shown to decrease the incidence of postcardiac surgical AKI.24 These guidelines recommend the avoidance of nephrotoxic agents, the close monitoring of serum creatinine and urine output, the avoidance of hyperglycaemia, the consideration of alternatives to radiocontrast agents and a close haemodynamic monitoring to maintain adequate CO, blood pressure and peripheral oxygen delivery. To reduce AKI incidence related to associated factors during prolonged CPB, the KDIGO guidelines have been implemented at our institution as a standard of care in all patients undergoing cardiac surgery.
Anaesthesia management of patients enrolled in the study is performed according to the hospital standard of care and using standardised protocols.36 Before entering the operating room (OR), patients are premedicated with 2–5 mg of midazolam. Prophylactic antibiotics (cefazolin 2–3 g or clindamycin 900 mg for patients allergic to cefazolin) are administered through a peripheral venous catheter (Introcan Safety IV Catheter, B Braun). These are given during the hour before the surgical incision. In the OR, ECG and peripheral oxygen saturation (SpO2) are continuously recorded. A radial arterial catheter (Arrow Seldinger Arterial Catheter) is placed for continuous blood pressure monitoring and blood sampling. Patients are then preoxygenated at an inspired oxygen concentration of 100% for 3 min. Anaesthesia is then induced with an intravenous bolus injection of 4–6 µg/kg of fentanyl and either propofol 2 mg/kg or etomidate 0.1 mg/kg. Rocuronium 1.2 mg/kg or cisatracurium 0.2 mg/kg are used for paralysis prior to orotracheal intubation. Medication doses are adjusted in elderly patients. After intubation, patients are connected to the anaesthesia machine, and mechanical ventilation is delivered in volume-controlled mode with a tidal volume of 6 mL/kg. Respiratory rate is titrated to maintain a partial pressure of carbon dioxide (PaCO2) between 35 and 45 mm Hg. The inspired fraction of oxygen (FiO2) is titrated according to the partial pressure of oxygen (PaO2), which is measured with arterial blood gas analysis. The TEE probe (X7-2t 3D ultrasound probe, Philips) is inserted by the cardiac-anaesthesia team for real-time intraoperative feedback to cardiac surgeons on (1) right and left ventricular function, (2) the mechanical stability and the function of repaired/replaced heart valves and (3) the detection of air emboli/clots. The neck, chest and abdomen are prepped and draped in a sterile fashion. A urinary catheter is positioned for hourly urinary output monitoring. To determine intraoperative and postoperative effects of NO on haemodynamics, after induction of anaesthesia, a PAC (Edwards Lifescience, Irvine, California, USA) is placed in all patients for continuous haemodynamic monitoring of central venous pressure (CVP), pulmonary artery pressure (PAP) and cardiac index (CI). Electrodes for SEDLine brain function monitoring (Masimo, Irvine, California, USA) are positioned on the forehead of each patient to assess the depth of anaesthesia by displaying frontal cortex electroencephalogram waveforms and measuring the patient state index (PSI).37 The level of anaesthesia is maintained by isoflurane administration (0.8–1.2 minimum alveolar concentration) and adjusted based on haemodynamics, while sustaining a PSI score between 30 and 50. Paralysis is maintained throughout the entire procedure by continuous infusion of rocuronium 4–10 µg/kg/min or cisatracurium 1–3 µg/kg/min. When clinically indicated, inotropes or vasopressors are administrated to maintain a mean arterial pressure (MAP) >65 mm Hg and a CI >2.2 L/min/m2.
The perfusion equipment utilised at Massachusetts General Hospital consists of Stockert S5 (LivaNova, Mirandola, Italy) heart–lung machines, CardioQuip (CardioQuip, Bryan, Texas, USA) cooler heaters, the LivaNova Xtra autologous cell salvage system and Medtronic HMS Plus Hepcon system (Medtronic, Minneapolis, Minnesotta, USA).
The CPB circuit is primed with 1600 mL Ringer’s lactate and recirculated for priming and air removal. Prior to cannulation, the patient is fully heparinised to a target activated clotting time (ACT) of no less than 400 s and a target heparin concentration of no less than 2.0 IU/mL. The loading dose of heparin is calculated by the Hepcon device and subsequent ACT and heparin concentration assays are performed on the same system. The cannulation strategy may be central, peripheral or a combination of both depending on the type of surgery (coronary/valve vs aortic), level of urgency, surgical approach (median sternotomy vs thoracotomy), patient-specific variables such as body habitus, previous cardiac surgical history, vascular disease and vascular anatomic anomalies. Most commonly, an arterial outflow cannula is placed in the distal ascending aorta and venous drainage is achieved by placing a multistage venous cannula in the right atrium via the inferior vena cava. The aortic root is typically cannulated with a catheter to deliver antegrade cardioplegia as well as to decompress the left ventricle. Retrograde catheters are commonly placed in the coronary sinus to maximise myocardial protection during the aortic cross clamp.
Once fully cannulated and prior to CPB, 500 mL–1000 mL of prime volume may be removed from the CPB circuit via retrograde autologous priming and/or venous antegrade priming to reduce the crystalloid burden on the patient on initiation of CPB. Once the patient is placed on CPB hypothermia is initiated to an extent dictated by type of surgery and surgeon’s preference. Mild hypothermia (temperature maintained between 32°C and 35°C) is most common for coronary and valve operations whereas circulatory arrest cases call for deeper hypothermia (temperature maintained between 18°C and 24°C).
Aortic occlusion is achieved by application of an aortic cross clamp and myocardial protection is achieved by administration of either Del Nido cardioplegia38 or a traditional 4:1 (blood:crystalloid) mixture at hypothermia into the aortic root (antegrade) and the coronary sinus (retrograde). Diastolic arrest is maintained by intermittent doses of cardioplegia every 20–30 min.
Blood flow rates while on CPB are maintained at a CI of 2.4 L/min/m2 or greater, a MAP of 65–75 mm Hg and a urinary output of greater than 0.5 mL/kg/hour. To achieve these MAP goals, phenylephrine may be titrated to effect and diuretics such as furosemide (10 mg bolus) may be given. Sweep gas flow and FiO2 are regulated to maintain a PaO2 of 150–250 mm Hg, PaCO2 of 35–45 mm Hg and a mixed venous oxygen saturation (SvO2) greater than 65%. Arterial and venous blood gases are drawn every 30 min and analysed by the hospital laboratory. Ultrafiltration with a LivaNova hemoconcentrator (LivaNova, Mirandola, Italy) may be performed with or without maintaining zero balance fluid administration for the purposes of normalising potassium levels (target K+ 3.5–5.5 mmol/L) and to increase haematocrit (HCT) (target HCT greater than 21%), respectively. In the event, ultrafiltration is inadequate to increase HCT to greater than 21% packed red blood cells (pRBCs) or red blood cells processed by intraoperative autotransfusion may be administered via the LivaNova Xtra autologous cell salvage system. Target blood glucose levels of 100–180 mg/dL are maintained by administration of intravenous insulin (bolus 10 IU and continuous infusion of 2 IU/hour) when the glucose level exceeds 180 mg/dL.
Once the procedure is complete, the heart and aorta are deaired and the aortic cross clamp is removed. Temporary epicardial pacing leads are placed, the surgical site is policed for bleeding and the patient is weaned from CPB. On satisfactory termination from CPB, the cannulas are removed and the patient is administered protamine sulfate to reverse heparin anticoagulation at a dose indicated by the Hepcon system. Full heparin reversal is confirmed by a heparin level assay and ACT measurement. Chest tubes are inserted and the sternum is approximated according to procedure and surgeon preference.
After surgery, patients are transferred to the cardiac surgical intensive care unit (CSICU). Standardised protocols for temperature, sedation, pain, glucose and haemodynamic management are followed. On CSICU arrival, active rewarming using a forced-air warming device is performed. Propofol 2 mg/kg/hour, ketamine 0.5 mg/kg/hour or dexmedetomidine 0.5–1.5 µg/kg/min are administered and titrated to achieve appropriate sedation. Pain control is achieved with the use of multimodal regimens (intravenous opioids, acetaminophen and non-steroidal antiinflammatory drugs). Within the first 72 hours, blood glucose levels are monitored every 4 hours and intravenous insulin may be used to maintain glucose levels between 100 and 180 mg/dL. Volume status and haemodynamic parameters are continuously monitored throughout the patient’s CSICU stay by arterial line and PAC. If needed, vasopressors are administered to achieve the following haemodynamic targets: CI >2.2 L/min/m2, MAP >65 mm Hg and SvO2 >65%. CI, pulmonary vascular resistance and systemic vascular resistance are recorded every 4 hours until test gas suspension. The pRBCs are transfused when Hb <7 g/dL.39 Urinary output is recorded hourly via urinary catheter. The ICU team may administer furosemide intravenously in the presence of oliguria only after optimisation of haemodynamic parameters, volume status and assurance of no urinary catheter blockage. No nephrotoxic agents (eg, intravenous contrast or hydroxyethyl starch products) are administered as recommended by the KDIGO guidelines.23
Gas delivery
After PAC insertion, patients are randomly allocated to the intervention group (80 ppm NO) or the control group (N2). Using commercially available tanks of N2 or NO (Airgas, Radnor Township, Pennsylvania) and volumetrically calibrated flowmeters, pure N2 (placebo) or 850 ppm NO gas in N2 is mixed with pure O2 or air. In the NO group, a final concentration of 80 ppm NO is obtained. Test gas administration starts at the onset of CPB and lasts for 24 hours. The test gas is administered via the CPB machine into the sweep gas of the CPB oxygenator by regulating the tank as follows:
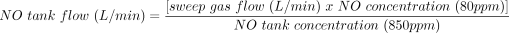
At the end of the CPB, once ventilation is resumed, test gas is delivered via the inspiratory limb of the anaesthetic or mechanical ventilator circuit, and thereafter via the mechanical ventilator in the CSICU. The concentration of NO is regulated by using a medical gas blender. When patients are extubated, they breathe test gas via a facemask or nasal prongs. NO and nitrogen dioxide (NO2) levels are monitored through in-line NO/NO2 sensors (Alphasense, Great Notley, Essex, UK). Met-Hb levels and SpO2 are continuously measured using a peripheral pulse co-oximeter (Masimo Rainbow Set Technology). Schemas illustrating methods of NO and N2 gas delivery and NO/NO2 monitoring both during surgery and in the CSICU are reported in figures 3 and 4.

Schema of the NO delivery systems in the operating room. (A) Figure illustrating how the NO or N2 is delivered into the CPB oxygenator. Tanks of pure N2 are used in the control group and tanks of 850 ppm NO in N2 are used in the intervention group. A ‘Y’ adaptor is inserted into the sweep gas input line leading to the oxygenator. This allows for the mixing of the test gas with the sweep gas (O2+medical air). This mixture is periodically monitored with an NO/NO2 analyser directly before entering the oxygenator. (B) Figure illustrating how the NO or N2 is delivered into the anaesthesia ventilator once ventilation has been resumed. The test gas is delivered by placing a ‘Y’ adaptor into the inspiratory limb of the circuit. The mixture is scrubbed of NO2 by a large volume scavenger containing calcium hydroxide and is periodically analysed with a NO/NO2 analyser before being inhaled by the patient. CPB, cardiopulmonary bypass; N2, nitrogen; NO, nitric oxide.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schema of the NO delivery systems in the cardiac surgical intensive care unit. (A) Figure illustrating how the NO or N2 is delivered through the mechanical ventilator at bedside in the intensive care unit. Tanks of pure N2 are used in the control group and tanks of 850 ppm NO in N2 are used in the intervention group. Test gas is blended with medical air and enters the air inlet of the ventilator. The high pressure O2 hose is directly connected to the ventilator. If there is any change of FiO2, the amount of NO/N2 delivered is regulated by the RT by adjusting the blender setting and the ventilator FiO2 setting, ensuring that the patient is still receiving the target concentration of 80 ppm NO. The mixture obtained is then scrubbed of NO2 through a large volume scavenger and a small volume scavenger placed in series on the inspiratory limb of the circuit. The final amount of NO and NO2 delivered is periodically analysed with a NO/NO2 analyser directly before the mixture is inhaled by the patient. (B) Figure illustrating how the NO or N2 is delivered into the high flow nasal cannula device. The test gas is delivered to the system by placing ‘Y’ adaptor before the humidifier. A commercially available blender mixes O2 and medical air and is regulated by the RT to reach the target FiO2. The flow of NO2 or N2 is titrated to reach the desired concentration 80 ppm NO or placebo. This mixture is then humidified and heated to a temperature of 34°C. FiO2, inspired fraction of oxygen; N2, nitrogen; NO, nitric oxide; O2, oxygen; ppm, parts per million; RT, respiratory therapist
Abrupt discontinuation of inhaled NO may lead to rebound pulmonary hypertension. This is characterised by worsening oxygenation and increased PAP, resulting in hypoxemia, systemic hypotension, bradycardia and right ventricular failure.40–42 For these reasons, after 24 hours, NO is weaned and discontinued while carefully monitoring haemodynamics using the PAC. NO weaning protocols at our institution consists of the following.
Decreasing NO to a half dose and maintaining this dose for 5 min.
Further decreasing NO dose to 5 ppm and maintaining this dose for 5 min.
Complete discontinuation of NO if no pulmonary hypertension rebound occurs and haemodynamics are stable for 10 min after NO gas suspension.
If at any time during the weaning protocol a patient experiences:
Worsening hypotension (systolic blood pressure, SBP <90 mm Hg) requiring the initiation of an intravenous infusion of vasopressor agents or, in patients already receiving vasopressors before NO weaning, a 50% increase of the dose of the vasopressors.
Worsening hypoxemia requiring an FiO2 increase of more than 0.2 or a positive end expiratory pressure (PEEP) increase of more than 5 cm H2O.
mPAP increase of more than 20% of the value measured before the initiation of the NO weaning.
A CI decrease below 1.5 L/min/m2 in the presence of a mPAP over 25 mm Hg.
A respiratory therapist (RT) resets the NO to the lowest level at which the patient was stable and notifies the principal investigator (PI.) and the CSICU attending physician.
Failure of NO weaning is established when one of the above complications occurs.
Blood and urine collection
To assess the extent of haemolysis and the renal-protective properties of NO administration, plasma samples for plasma free Hb, NO consumption, NO metabolites and serum creatinine measurements are collected immediately before starting the surgical procedure, 15 min after the end of CPB, 24 hours after surgery and 48 hours after surgery.15 17 43–45 Additional blood samples for serum creatinine measurements are collected once a day for 7 days following the cardiac procedure or until discharge if the patient is discharged before day 7. Finally, a blood sample for serum creatinine is collected 6 weeks after surgery. Five urine samples are collected for urinary biomarkers of kidney injury (immediately before starting the surgical procedure, 15 min after the end of CPB, and at 24 hours, 48 hours and 6 weeks after surgery).46–48 After centrifugation, all specimens are coded, snap frozen with liquid nitrogen and stored on designated shelves in a −80°C freezer.
Outcomes
Primary outcome
The primary endpoint of this therapeutic trial is to determine whether there is a difference in AKI incidence between the control group (receiving N2) versus the intervention group (receiving NO). AKI is defined by KDIGO criteria as an abrupt (within 48 hours) reduction in kidney function correlated to an absolute increase in serum creatinine of 0.3 mg/dL or more (≥26.5 μmol/L) or a percentage increase in serum creatinine of 50% or more (1.5-fold from baseline) at any time during the first 7 days after surgery or a urinary output <0.5 mL/Kg/h for >6 hours49.
Secondary outcomes
Secondary renal outcomes include:
AKI severity using the KDIGO stages49:
Stage 1: serum creatinine increase ≥26.5 μmol/L (≥0.3 mg/dL) or increase to 1.5-fold to twofold from baseline or urinary output <0.5 mL/kg/hour for 6 hours.
Stage 2: serum creatinine increase more than twofold to threefold from baseline or urinary output <0.5 mL/kg/hour for 12 hours.
Stage 3: serum creatinine increase more than threefold from baseline or serum creatinine ≥354 μmol/L (≥4.0 mg/dL) or urinary output <0.3 mL/kg/hour for 24 hours or anuria for 12 hours or a need for renal replacement therapy (RRT).
Requirement for RRT following AKI during hospitalisation and at 6 weeks, 90 days and 1 year after surgery.
MAKE at 6 weeks after surgery. MAKE is a composite outcome of death, new RRT and worsened renal function (defined as a 25% or greater decline in eGFR).50
Secondary non-renal outcomes include:
Sequential organ failure assessment (SOFA) score during ICU stay.51
Prolonged cardiovascular support defined as the need for vasopressors and inotropic agents, a balloon pump or a ventricular-assist device for more than 48 hours after cardiac surgery.
Maximum hourly vasoactive-inotropic score (VIS) for the first 7 days after surgery and duration of vasopressors and inotropic agents support. VIS is calculated as dopamine dose (mcg/kg/min)+dobutamine dose (mcg/kg/min)+100×epinephrine dose (mcg/kg/min)+10×milrinone dose (mcg/kg/min)+10 000×vasopressin dose (units/kg/min)+100×norepinephrine dose (mcg/kg/min)+10×phenylephrine dose (mcg/kg/min).52 53
Duration of mechanical ventilation (hours).
ICU and hospital length of stay.
Exploratory outcomes
Severity of AKI using urinary biomarkers of kidney injury.46–48
AKI incidence and severity related to baseline characteristics of enrolled patients such as the presence of eGFR <60 mL/min at baseline, mPAP at baseline, cardiovascular risk factors associated with endothelial dysfunction, the type of surgical procedure, European System for Cardiac Operative Risk Evaluation (EuroSCORE II)54 and intraoperative course (ie, duration of CPB, duration of aortic cross-clamp, levels of plasma free Hb, levels of NO consumption and NO metabolites) that may impact study results.
Delirium assessed using the confusion assessment method for the ICU (CAM-ICU) within the first 7 days after surgery or until ICU discharge.55 56
Quality of life at 6 weeks, 90 days and 1 year after surgery assessed using the Katz Index of Independence in Activities of Daily Living (ADL) and the Patient-Reported Outcomes Measurement Information System global health questionnaire.57–59
Safety outcomes
Intrahospital mortality and mortality at 6 weeks, 90 days and 1 year after surgery.
Non-fatal stroke during hospitalisation and at 6 weeks after surgery.
Perioperative and non-perioperative non-fatal myocardial infarction.60
Postoperative bleeding calculated as the sum of blood loss through thoracic drains from the moment of chest closure over a period of 24 hours.
Need for blood transfusions or transfusions with blood products.
Postoperative infections (eg, pneumonia, wound infection, endocarditis, central line infection, urinary tract infection, sepsis).
Cardiac arrhythmias and other non-cardiac postoperative complications (eg, hepatobiliary disorders, pneumothorax, pleural effusion, vascular disorders).
Patients requiring a decrease of NO delivery due to Met-Hb >5%.
Sample size planning
The sample size needed for this trial is calculated based on the primary endpoint: the reduction of AKI incidence in the NO treated group (intervention group) compared with the N2 group (control group). In a RCT conducted at the University of Muenster (Germany), Meersch et al showed that implementation of a KDIGO CT surgery ‘bundle’ as recommended by the KDIGO guidelines successfully reduced the incidence of CPB-associated AKI from 71.7% to 55.1%.24 At Massachusetts General Hospital (MGH), the use of the KDIGO guidelines is part of standard care and the CPB-associated AKI rate is similar (55% as observed in a 1-year chart review performed by the investigators for surgery requiring CPB >90 min (data not shown)). In a prior trial we conducted in China, we found a 22% relative risk reduction in the incidence of AKI and a 42% relative risk reduction of stage 3 CKD at 1 year in the NO treated group.17 In this study, we estimate a greater reduction in the incidence of AKI (35% relative risk reduction) because we anticipate that an American population with endothelial dysfunction will benefit more from NO treatment. Thus, in the NO group, the incidence of AKI is expected to decrease from 55% to 35.75%. Using a Fisher’s exact test to compare proportions, the sample size needed to detect a difference, assuming a two-sided type I error of 0.05 and a power of 0.8, is 114 patients per group. In order to account for possible dropouts, we have increased our sample size by 10%. The enrolment plan is 250 patients (125 patients in the NO group and 125 patients in the N2 group).
Recruitment
This study is performed at MGH in Boston, Massachusetts, USA. All inpatients and oupatients undergoing a scheduled procedure are screened for eligibility. If a patient is excluded, the reasons are noted on a screening log. To obtain consent from scheduled patients, the details of the study are given during the preoperative visit at the Cardiac Surgery Preoperative Clinic, along with a flyer overviewing the trial protocol and a copy of the consent form. For patients admitted to the hospital, study details are given at least a day prior to their cardiac procedures. On the day of surgery, prior to the initiation of any study procedures, the patient’s written consent is obtained by a licensed physician who is also a member of the study staff. Consent is obtained exclusively from the patient and not from a surrogate. Patients who choose not to participate in this study receive standard care according to the surgical procedure. After consent is obtained, a de-identified code is assigned to the patient and registered on a dedicated enrolment log.
Randomisation and treatment allocation
To ensure balance between study groups with respect to the likelihood of receiving NO after surgery, after the insertion of a PAC, the patients are stratified by preoperative mPAP (ie, mPAP <30 mm Hg or mPAP ≥30 mm Hg) then allocated into the two study groups. The randomisation sequence is created by an independent statistician using Stata Statistical Software: Release V.14 and then uploaded on the MGH Research Electronic Data Capture (REDCap) application.61 A predetermined block randomisation method (fixed block size of 10) is used to ensure equal distribution of participants to treatment arms. A physician with no clinical involvement in providing care for the enrolled patients will conduct the randomisation procedure throughout the study by using the randomisation function provided by REDCap. Thus, the randomisation is conducted without any influence from clinicians providing care for the patients or from statisticians or outcome assessors.
Blinding
The NO and N2 tanks and the gas delivery systems in the OR and at the bedside are masked and cannot be distinguished on the basis of appearance. This allows to keep participants, clinicians and investigators blind to the assignment group. For safety and gas monitoring, the clinician administering the test gas remains unblinded to the treatment. This unblinded clinician is solely responsible for gas tank preparation and test gas delivery and monitoring. Assessors who are blind to the treatment allocation conduct assessment of study outcomes. Blinding is maintained until 1 year follow-up. The allocation of patients enrolled in the study will not be disclosed before completion of the study (250 patients enrolled). To maintain the overall quality, legitimacy and integrity of the clinical trial, unblinding of the test gas may occur only in critical circumstances when the responding physician prescribes initiation of NO. In this circumstance, the PI. fully documents and explains the reasons for unblinding in a report to the Institutional Review Board (IRB).
Data analysis
All trial outcomes will be evaluated using an intention-to-treat analysis plan. Hypothesis testing will be performed using a two-sided significance level (type I error) of α=0.05. Sensitivity analyses per group of treatment will also be performed and compared with the intention-to-treat analysis results. The incidence of AKI (primary endpoint) and postoperative adverse events after prolonged CPB will be reported in terms of relative risk with 95% CI in the treatment versus the control group. The median differences (NO group vs control group) with 95% CI will describe differences in continuous perioperative characteristics and continuous postoperative outcomes.
An unadjusted analysis of the primary endpoint will be performed by means of Fisher’s exact test comparing incidence of AKI in the NO group against the control (placebo) group. Multivariate logistic regression will be performed to determine if treatment with NO reduces incidence of AKI after adjusting for eGFR, age, sex and EuroSCORE II.
Secondary and exploratory endpoints will be addressed using appropriate tests for each outcome. Categorical variables will be compared between the two groups using Fisher’s exact test or Χ2 as appropriate. Continuous variables will be compared between the two groups using Student’s t-test or a Mann-Whitney U test as appropriate. The change in the laboratory markers over time and between the two groups will be tested with a mixed effects modelling. Differences among the groups at each timepoint will be tested using an unpaired Student’s t-test or a Mann-Whitney U test as appropriate.
Interim analysis
An interim analysis by a Data Safety Monitoring Board (DSMB) is planned upon reaching 50% of the study population. The DSMB is composed of three independent, multidisciplinary experts (online supplementary material: Data and Safety Monitoring Board section). An independent statistician will perform the interim analysis. The statistician will report the results of the data analysed to the DSMB in a closed session. The DSMB will have access to data collected during the hospital stay and during the follow-up visits, including maintenance of patient confidentiality throughout the study. The DSMB may recommend terminating the study if at the interim analysis a significant increase of intrahospital mortality, mortality at 6 weeks, AKI or need for RRT in either group (N2 or NO) is detected.
Supplemental material
Data management
All data collected for the trial are entered into the MGH REDCap application.61 Preoperative, intraoperative and postoperative data are prospectively collected from the patient’s chart until discharge. Follow-up data are collected by interviewing patients at 6 weeks, 90 days and 1 year after surgery.
Data access is restricted and granted by the PI only to authorised members of the study team. The PI also assigns specific privileges to members of the study team for data import/export. Quality and integrity of the data collected are optimised by using software properties such as logic checks and validation of data fields (ie, reference range, valid or invalid values). Warnings and error messages alert the co-investigator of missing data or data entries that do not match the requirements set up in the data-entry field. Also, the co-investigators manually perform weekly checks to ensure data consistency. During these periodical checks, all errors detected by the study team related to data collection and data entry are summarised in a data query report. The report is sent to the PI following the enrolment of every five patients. It is the responsibility of the PI to check the original forms for inconsistency, make corrections by modifying the original forms when necessary and enter a response to the query. Strict confidentiality is maintained by the research team at all times. All forms are and will be kept in a secure, locked cabinet with limited access for at least 5 years after study completion.
Trial management
Study team physicians are responsible for screening all inpatients and outpatients undergoing a scheduled procedure. The number of eligible, consented, enrolled and randomised patients is recorded in addition to the reasons for non-participation in the trial.
During their hospital stay, patients are closely monitored and all outcomes are recorded. For this reason, no missing endpoints are expected. After discharge, all patients have a scheduled follow-up visit with the cardiac surgeon 6 weeks after surgery. The day before their appointment, the patient is contacted by a member of the study staff to remind them that a member of the team will collect research data during their visit. This is done by a phone call (or email if unreachable by phone). At the end of the visit with the cardiac surgeon, blood and urine samples are collected for kidney function assessment and a questionnaire regarding medical and surgical complications and quality of life is given. Patients are then called at 90 days and 1 year after surgery to evaluate mid-term and long-term outcomes. To avoid missing follow-ups, a minimum of three calls are attempted by the study team. Calls are made at different times and dates in an attempt to increase the probability of contacting patients. Phone numbers provided by the patient on the screening day are used. If the study team is still unable to contact the patient despite the several phone call attempts, a letter is sent to the home address provided by the patient at their initial screening visit. If all attempts do not provide contact with the patient, a member of the study team may call the patient’s primary care physician or another healthcare provider to obtain information regarding the patient’s condition (eg, deceased). Based on trials previously performed in the Department of Surgery at the MGH, we expect a loss to follow-up of 10%, 15% and 20% at 6 weeks, 90 days and 1 year, respectively.
Trial risks
Due to the instability of NO, there are risks associated with its use that must be considered. NO reacts slowly with oxygen to form NO2, which may cause airway inflammation and damage to lung tissues.62 Moreover, NO oxidises ferrous Hb to form Met-Hb, which is unable to transport and release oxygen to tissues. However, cyanosis in healthy patients does not appear until Met-Hb levels are 15%–20%.63 The binding of NO to Hb is a rapidly reversible reaction, with a half-life of 15–20 min after NO discontinuation. The side effects and adverse events related to NO delivery are well reported in the literature. In table 1, we summarised eight clinical trials that recorded and reported adverse events associated with NO gas in newborn, paediatric and adult populations. Based on the present literature and Food and Drug Administration reports,64 the risks of breathing NO at 80 ppm for 24 hours are minimal when Met-Hb levels and NO/NO2 delivery levels are carefully monitored.65 66 To improve safety, in the present trial, NO is administered and monitored by trained respiratory therapists. The IRB for this study requested monitoring and maintaining levels of NO2 breathing below 5 ppm. We previously tested and found that our delivered circuits do not exceed delivering 1.5 ppm of NO2 when 80 ppm of NO gas is delivered at 90% of FiO2. Met-Hb is continuously monitored by non-invasive co-oximetry. If Met-Hb levels exceed 5% of circulating Hb, the concentration of NO delivered is halved from 80 ppm to 40 ppm and closely monitored until a reduction occurs. If Met-Hb levels persist above 5%, NO is progressively halved until a reduction below 5% occurs.
Summary of prior selected studies that reported adverse events related to NO administration
Criteria for patient discontinuation
The criteria for patient discontinuation from the study are as follows.
Voluntary discontinuation by a patient.
Exiting the protocol for safety reasons based on the judgement of the clinical or research staff. Specific safety reasons include: (1) acute worsening of hypotension defined as a decrease in MAP of >20 mm Hg that is not attributable to other causes (such as hypovolemia, haemorrhage or sepsis), but is generally due to worsening of left ventricular failure, (2) sudden worsening of hypoxemia defined as a decrease of SpO2 to <80% while breathing 100% oxygen and not attributable to other causes (such as pulmonary oedema, ARDS or pulmonary embolism) and (3) an increase in NO2 levels >5 ppm from baseline.
Patient and public involvement
Patients were not directly involved in the study design and recruitment, and did not contribute to the development of the research questions and outcomes. At the end of the trial, on request, results will be provided to the study participants and/or their family members and then published on clinicaltrials.gov.
Enrolment to date
As of May 2019, 161 patients have been enrolled in the study. Assuming that we recruit about eight patients per month, we anticipate completing the recruitment of 250 patients by May 2020.
Ethics and dissemination
The current trial was approved by the International Review Board (IRB) in August 2016. Further amendments of the study protocol have been performed and clinical trials.gov has been updated accordingly. Analysis of the outcomes of the study (primary, secondary and exploratory outcomes) will be performed when the enrolment of 250 patients is complete. These results will be published in a primary paper, which will be directed to a peer-reviewed journal and may be presented as a poster and/or through oral communication at scientific conferences focused on cardiac surgery, nephrology, anaesthesia and critical care medicine.
Conclusion
This trial is evaluating whether 24 hours administration of NO gas reduces the incidence of AKI after prolonged CPB in patients with signs and symptoms of endothelial dysfunction. The results obtained by this trial may also represent further progress of the scientific community in evaluating the systemic beneficial properties of exposing plasma to NO gas.
Acknowledgments
The clinical perfusionists of the Department of Cardiac Surgery, Perfusion Services, Massachusetts General Hospital (Boston, Massachusetts, USA). The respiratory therapy staff, the nursing staff and the physicians of the Department of Anesthesia, Critical Care and Pain Medicine, Massachusetts General Hospital , USA. The nursing staff and the physicians of the department of Cardiac Surgery, Massachusetts General Hospital (Boston, MA, USA). The nursing staff of the pre-admission testing area, Massachusetts General Hospital (Boston,MA, USA). Francesco Nordio, PhD (Research Associate in Medicine, Department of Medicine, Brighamand Women’s Hospital, Boston, MA, USA) for his role of independent statistician of this trial. Kelly Larson, RN, and Christopher Chenelle, BA, for language editing.
References
Footnotes
Contributors Authorship for this trial will be given to key personnel involved in trial design, personnel training, recruitment, data collection, statistical plan and data analysis. There are no publication restrictions. LB, WMZ, JVB, TBT, NR, EAB, TMS, DF, ER, FM and FI were responsible for conceptualising trial design. LB managed patient safety protocol. LB, FM, SS, FZ, CX, GL and TSL are responsible for recruitment, enrolment and data collection. RM, HZ and EAB are responsible for power calculation, statistical plan and data analysis. LB, KS, SM, WDR, PW and RMK trained personnel for the clinical trial and built systems for nitric oxide delivery and monitoring. All authors have critically revised the study protocol and approved the final version. All authors agree to be accountable for the accuracy and integrity of all aspects of this trial.
Funding Department of Anesthesia, Critical Care and Pain Medicine (Massachusetts General Hospital, Boston, Massachusetts, USA) and the National Institutes of Health (National Heart, Lung, and Blood Institute K23 HL128882-01A1).
Competing interests FM and LB salaries are partially supported by NIH/NHLBI 1 K23 HL128882-01A1. JB is co-inventor on patents that are assigned to Partners Healthcare. RMK is a consultant for Medtronic and Orange Medical and has received research grants from Medtronic and Venner Medical.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.