Article Text

Abstract
Introduction Existing interventions for people with Parkinson’s disease (PwP) often fall short in addressing gait disturbances and falls, impacting their quality of life. The CUE1 non-invasive medical device, along with its updated version, CUE1+, offers vibrotactile stimulation with cueing. The device shows promise in alleviating motor symptoms and reducing falls based on early user testing and a 9-week pilot study. This study aims to assess the usability, safety, tolerability and effectiveness of CUE1+ in improving Parkinson’s symptoms compared with a sham device over a 12-week period.
Methods and analysis This multicentre, phase II double-blind randomised controlled trial will recruit 50 PwP from Barts Health and Homerton NHS Hospitals, enrolling them at Queen Mary University of London. Participants, diagnosed with idiopathic Parkinson’s, aged 18+ and providing written consent, will be randomly assigned to either the experimental group (CUE1+ device) or control group (sham device). The primary outcome is the device usability over 12 weeks. Measures include the recruitment, compliance and dropout rates, and safety/tolerability which will be collected through a participant clinical diary at baseline (week 0) and follow-up (week 13). Effectiveness will be evaluated at the same time points using movement tests (MDS-UPDRS Part III, Functional Gait Assessment, Timed Up and Go in isolation and with dual tasking and two keyboard-based typing tests—Bradykinesia Akinesia Incoordination and Digital Finger Tapping), with video recordings. Participants will wear a Parkinson’s KinetiGraph wristband to monitor symptoms at home continuously for 12 weeks and collect real-world data. Patient-reported outcomes will be collected at baseline and follow-up and include MDS-UPDRS Part I, II and IV, Activity-specific Balance Scale, Pittsburgh Sleep Quality Index, Hospital Anxiety and Depression Scale, Fatigue Symptom Scale and Parkinson’s Disease Questionnaire-39.
Ethics and dissemination The study has received ethical approval from London-Dulwich Research Ethics Committee (reference: 23/PR/1526). Findings will be submitted for peer-reviewed publications.
Trial registration number NCT06174948.
- Wearable Electronic Devices
- Gait
- Protocols & guidelines
- Parkinson-s disease
- Randomized Controlled Trial
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This 12-week double-blind phase II (pilot) randomised controlled trial will allocate 50 people with Parkinson’s disease (PwP) to either active CUE1+ or sham device group.
PwP will be identified from Barts Health and Homerton NHS Trusts, with informed consent and participant flow managed according to Consolidated Standards of Reporting Trials and Standard Protocol Items: Recommendations for Interventional Trials guidelines.
This study does not accept PwP who are receiving interventions outside standard Parkinson’s care or are not on stable Parkinson’s medication for at least 3 months.
This pilot study is not powered to determine whether CUE1+ improves symptoms in PwP.
Data will be securely managed in accordance with the UK Data Protection Act, and findings will be disseminated through peer-reviewed publications, conferences and targeted updates to NHS sites and Parkinson’s support groups.
Introduction
Parkinson’s disease (PD) is a growing global issue, affecting around 153 000 people in the UK.1 It is marked by the degeneration of dopaminergic neurons in the substantia nigra, leading to movement symptoms including bradykinesia, rigidity, tremor, postural instability and freezing of gait (FOG).2 These symptoms severely impact mental health and quality of life (QoL).3 Non-motor symptoms, such as sleep disturbances, sensory impairments, fatigue, anxiety and depression, can be equally or more debilitating.2 3
Despite pharmacological treatments, many people with Parkinson’s (PwP) still experience symptoms, and dopaminergic therapy often causes side effects like akinesia, dyskinesia and psychosis.4 5 It can also exacerbate postural instability, raising the risk of falls,4 a major cause of morbidity and mortality in PwP. As a result, current interest has shifted towards cueing and vibrotactile stimulation to ease motor symptoms in PD.6 7 These interventions, delivered through non-invasive devices, have been shown to improve mobility, balance, gait disturbances including FOG and QoL at low cost. They are also recognised for their safety and tolerability among PwP.6–8
The CUE1 is a non-invasive medical device that is worn on the sternum and provides cueing with focused vibrotactile stimulation. Wearing the CUE1 has resulted in positive effects on movement symptoms, balance, walking, FOG and fall risk.8–10 A pilot interventional study conducted at Queen Mary University of London (QMUL) (reference number: 23/PR/1526) demonstrated high recruitment (83%) and compliance (100%) rates, with PwP expressing comfort and willingness to continue using the device long-term, beyond the study period.7 The CUE1 device demonstrated immediate and cumulative positive effects on motor symptoms, gait and balance, as well as a reduction in fall risk in PwP from diverse ethnic backgrounds (eg, white, mixed, Asian and black). Additionally, it showed positive effects on motor fluctuations and non-motor symptoms, such as sleep, over the course of the 9-week trial. However, the study lacked a control group, a sham device and assessor blinding, limiting the ability to fully assess its impact, technology readiness and regulatory decision.
Therefore, this study aims to test the enhanced CUE1+ in a larger, diverse group with a control group using a sham device. The CUE1+ device has the same intervention settings as its previous version, the CUE1 device, but the battery life has been extended. The 12-week study will assess the usability, including safety and tolerability, of the CUE1+ device as well as its effects on motor symptoms, balance, gait speed, FOG, fall risk and QoL in clinical and real-world settings. Participants will wear the Parkinson’s KinetiGraph (PKG)11 12 wristwatch to track symptoms continuously for 12 weeks while continuing their daily activities and complete patient-reported outcomes on movement, balance, sleep, psychological status, fatigue and QoL. This real-world data will offer insights beyond clinical assessments, capturing more relevant, day-to-day experiences for PwP.
Methods and analysis
Trial design
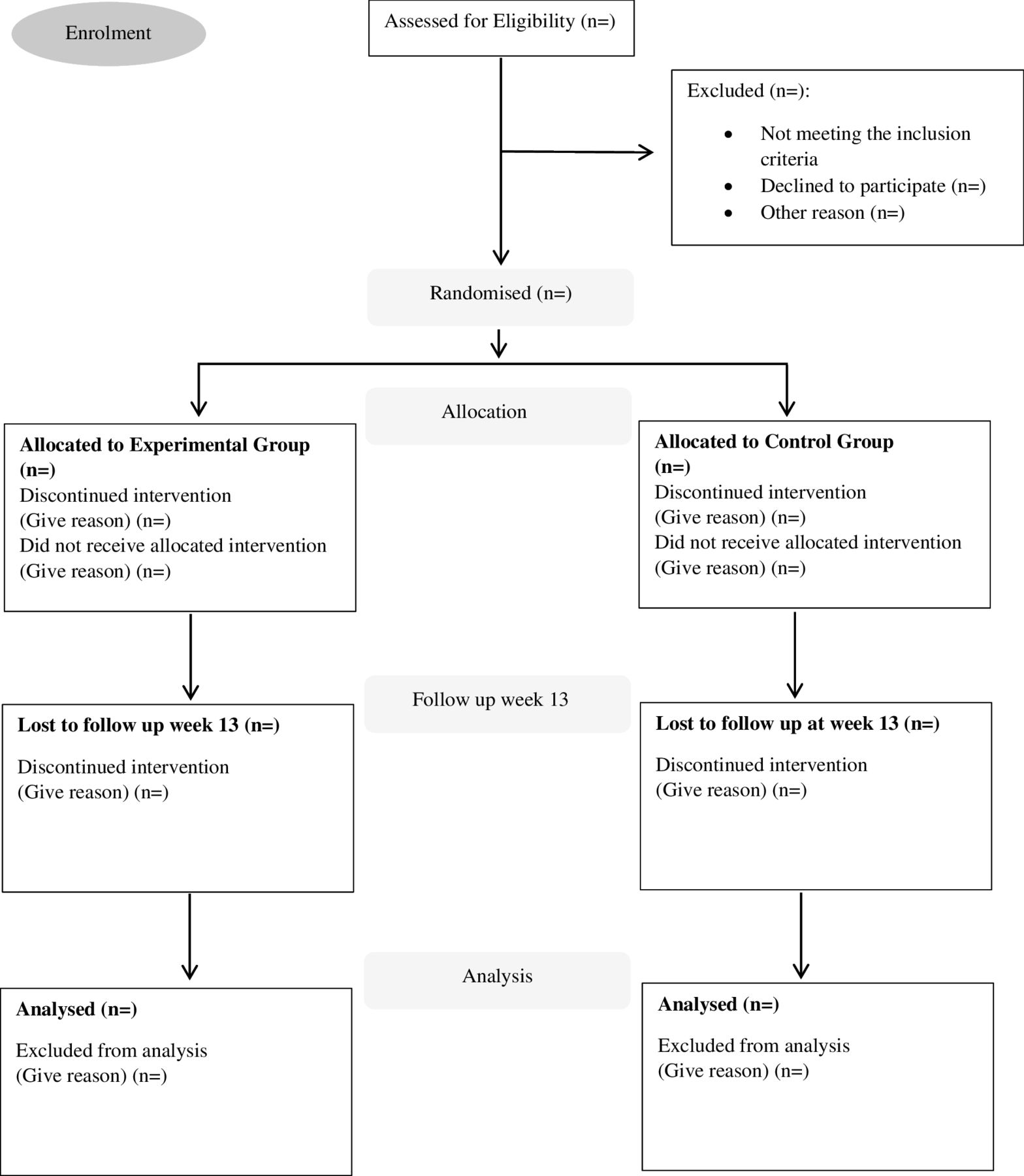
This 12-week multicentre double-blind phase II (pilot) randomised controlled trial (RCT), conducted by the Centre for Preventive Neurology at QMUL in collaboration with Barts Health National Health Service (NHS) Trust and Homerton NHS Hospitals, will randomly allocate 50 PwP to experimental or sham device group. The study is an extension of a feasibility study7 conducted by the same research team which received ethics approval from London-Dulwich Research Ethics Committee, London (reference: 23/PR/1526). The feasibility study and this extension are registered with ClinicalTrials.gov and comply with the Declaration of Helsinki. Informed consent will be obtained before data collection (see online supplemental material 1 for participant consent form). Participant flow will adhere to the Consolidated Standards of Reporting Trials statement guidelines (figure 1). The study protocol follows the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT)13 and the SPIRIT reporting guidelines.14
Supplemental material

CONSORT diagram to demonstrate participant flow through the CUE1+ study. CONSORT, Consolidated Standards of Reporting Trials.
Participant identification and recruitment
Participants identification and recruitment started in November 2024 from the Neurology Department at Barts Health NHS and the Care of the Elderly Department at Homerton Healthcare NHS Foundation Trust. Assessments take place at the Centre for Preventive Neurology, Wolfson Institute of Population Health, QMUL. Neurologists (AZ, DG, AN and CS), geriatricians (CQ, TB), senior physiotherapists (KR, VA), PD nurse (CB) and other members of the research team (EB, EC and KD) identify potential participants. A senior neurological physiotherapist (VA), trained for this study, manages consent and data collection. VA will remain blinded to the intervention. This blinded trial is expected to finalise data collection by June 2025. Following this, an additional 6 months will be required for data analysis, dissemination, manuscript preparation and submission to a scientific journal.
Inclusion criteria are clinical diagnosis of idiopathic PD,15 adults over 18 years old, willing to participate and provided written consent after reading the participant information sheet. Participants are excluded if they have a diagnosis that might affect their movements, balance and ability to carry out the trial independently. These include any neurological disorders other than PD, including atypical parkinsonism, osteoarticular problems, visual disturbances, audio-vestibular disorders and clinical diagnosis of dementia or Alzheimer’s disease. We also exclude those PwP receiving other cueing or vibrotactile stimulation or device interventions or therapeutic interventions apart from that provided as part of the standard care for PD-related movement symptoms and/or who were not on stable PD medication for a minimum of 3 months. Moreover, exclusion also applies to individuals with (a) implanted metallic or electronic devices, (b) known hypersensitivity to vibrotactile stimulation and/or (c) skin conditions or open wounds near the CUE1+ application site (eg, sternum).
Randomisation
To ensure impartiality, randomisation must be concealed from trial clinicians.16 Randomisation was conducted using an online randomisation service (sealedenvelope.com) to ensure a fully concealed allocation sequence. A block randomisation approach was implemented with varying block sizes (2, 4, 6) to maintain balance across the study arms and reduce predictability. Multiple random sequences (1–4) were used, along with block identifiers (1–14) to track allocation while preserving blinding.
One of the researchers (CS) oversees the randomisation process and has access to the allocation codes, while both the participants and the researcher conducting assessments (VA) remain blinded. Randomisation codes are securely stored and not disclosed to anyone except the designated unblinded researcher (CS) responsible for managing safety monitoring and adverse event reporting.
Blinding
This double-blind phase II RCT compares a technological intervention (CUE1+) with a sham device. All the participants and the research team member who performs the assessments (VA) as well as the statistician are unaware of group assignments. They will remain blinded until the last participant has completed the study and the results are analysed. Any unblinding will be documented with details on how and why it occurred. The Principal Investigators and research team do not anticipate the need for emergency unblinding.
Intervention
Experimental intervention: CUE1+ device
The experimental group will use the CUE1+ device with its original settings (figure 2) daily for 12 weeks. The CUE1+, developed by Charco Neurotech, is a CE-marked and MHRA-registered device that combines low-frequency auditory cues with high-frequency vibrotactile stimulation (see table 1 for specifications). Participants will wear the device for 8 hours each morning, starting 1 hour after taking their PD medication, with pre-set settings (80% vibration strength, 800 ms pulse length, 800 ms rest length) proven effective in prior studies.7–10 The device is attached to the sternum using a dermatologically approved adhesive patch. All participants will receive training on device and charger use and adhesives, along with a support booklet. They will also be able to contact the research team for any queries or technical support as needed. At the end of the study, participants may keep the CUE1+ for long-term use or return it.

{kind=link}
{kind=link}
The CUE1+ device may help improve motor and non-motor symptoms in people with Parkinson’s using a non-invasive approach. It is a discreet device (40 mm diameter, 11 mm height, 17 g) and attaches to the sternum with dermatologically tested adhesive patches. Water-resistant but not waterproof, it should be removed before showering. When active, it uses a silent motor to provide focused vibrotactile stimulation, following a specialised pattern based on extensive user testing and clinical research. It combines low frequency auditory cueing with high frequency focused vibrotactile stimulation through its distinct wave shape and frequency patterns. The CUE1+ sham device has the same exact appearance, but its treatment settings have been de-activated for the purposes of the study.
Specification of CUE1+ device
Control group
The control group will use a sham device for 12 weeks. This device is identical to the CUE1+ but with vibration strength set to 0%, pulse length at 0 ms and no changes throughout the study. Participants will wear it for 8 hours each morning, starting 1 hour after taking their PD medication, and will be informed that the device is active, though they may not feel any vibrations as the study aims to study a silent interventional setting. The device will be attached to the sternum using an approved adhesive patch. When participants complete the study, they will be informed that their device was sham and asked to return it. Their sham device will then be replaced with a new, active CUE1+ device, which participants in this group will be gifted and can use long-term as intended.
Training and a support booklet will be provided to both groups. Please refer to online supplemental material 2 for additional explanatory instructions that will be provided to all participants, aimed at supporting them with their device.
In both groups, patients are advised to carry on taking their regular medication. The exclusion criteria of the study highlight also that they need to be on a stable dose during the trial. If this is not possible, they will be withdrawn from the study.
Assessments and outcomes
Assessments will occur at baseline (week 0) and follow-up (week 13). Each participant will spend about half a day completing the movement assessments and the questionnaires. Breaks will be given as needed for each participant. Demographic data including age, gender, ethnicity, disease severity (Hoehn & Yahr scale; H&Y),17 disease duration, hand most affected by PD, hand dominance and cognitive status (Montreal Cognitive Assessment Tool; MoCA) will be collected. The MoCA is a screening tool for cognitive impairment, assessing various domains like attention, memory and executive function.18 A score above 25/30 suggests normal cognition. The order of assessments (eg, demographic data collection, questionnaires, physical assessments) will be performed in random order to minimise the fatigue bias. The sequence of events for all enrolled participants can be found in table 2.
Order of events for all participants enrolled in the project
The primary outcome is the usability of CUE1+ over 12 weeks in home settings, focusing on recruitment rate, compliance (usage duration and frequency), dropout rates and safety/tolerability. Safety and tolerability are assessed by total usage time, adverse reactions, attachment/removal issues, average comfort (5-point scale) and malfunction incidents. All will be explored through the participant clinical diary (online supplemental material 3). The participant clinical diary includes four sections: (a) symptom severity; (b) symptom frequency; (c) balance and falls (balance, gait, ‘near falls’, falls and QoL impact rated 0–10); (d) questions on CUE+ which will cover usability, safety, tolerability and effects on symptoms, independence, QoL, mood, social interaction, fear of falling and balance confidence.
The movement assessments using the MDS-UPDRS III,19 Functional Gait Assessment (FGA),20 TUG,21 TUG with dual tasking (DT) (eg, a cognitive numeracy task; counting backwards from 100 in 7s),22 Bradykinesia Akinesia Incoordination (BRAIN) tap test23–25 and Digital Finger Tapping26 will be conducted at baseline before using the device and in the end after the 12-week intervention is completed. Video recordings will be held for MDS-UPDRS III, FGA, TUG and TUG with DT for participants who will agree in the consent form to be recorded. All tests will be performed in the ON-medication state defined by specific timing—45–60 min after taking the medication.
Participants will be asked to wear the PKG11 12 wristwatch to track their movement for 12 weeks, starting from day 1 during the baseline assessment and finishing after the follow-up assessment at week 13. Participants will wear the PKG watch while carrying out their regular activities of daily living and at night during the whole trial, only taking it off when this needs to be charged for approximately 2 hours every 6–7 days. The main data points from the PKG system include median bradykinesia score, median dyskinesia score, fluctuation dyskinesia score, per cent (%) with tremor, percent time immobile, average steps per day, percent time in sleep, uninterrupted sleep duration (median in min) and percent sleep quality.
Patient-reported outcomes for motor fluctuations, balance confidence, non-motor symptoms, sleep, psychological state, fatigue and QoL will be assessed using MDS-UPDRS I, II and IV,19 Activity-specific Balance Scale,27 Pittsburgh Sleep Quality Index,28 Hospital Anxiety and Depression Scale,29 Fatigue Severity Scale30 and Parkinson’s Disease Questionnaire-39,31 respectively (see online supplemental material 4 for description of motor and patient-reported outcomes).
Participants will be unblinded when the trial finishes. At that time, participants in both groups will also complete the participant satisfaction form (see online supplemental material 5) on using their CUE1+ device.
Data management
Data management will adhere to the UK Data Protection Act 2018 and GDPR 2018, with personal data retained for 25 years as best practice. Essential trial documents will be securely kept with the Trial Master File and Investigator Site Files and stored online by QMUL, with study documentation retained by the sponsor and access restricted to authorised personnel. The integrity of collected data that have been inputted into the study database will be assessed by taking a random sample (10%) from participant files after the completion of data collection and comparing with the Case Report Form (CRF). If there is less than 98% agreement between the CRF record and inputted measures, all data will be checked and rectified. The rectified database will be saved under a new filename (eg, studydatabase_rectified_date_version) and all changes made to the database will be logged.
Statistical considerations
Researchers will conduct this phase II double-blind (pilot) RCT, and the statistician will provide a statistical plan for a future fully powered trial based on the results of this study. They will analyse data to estimate sample size, assess the effect of CUE1+ intervention and compare group changes while checking assumptions (eg, χ2) for accuracy.
Method of analysis
Statistical analysis will use IBM SPSS V.29. Descriptive statistics will summarise the data, while χ2 and Mann-Whitney U tests will assess group differences. Normality will be checked via the Shapiro-Wilk test, histograms and Q-Q plots. Data will be presented as mean±SD, with minimum, maximum and median values. Paired t-tests will compare pre- and post-intervention scores and the effect size for Mann-Whitney U tests will be calculated by dividing the standardised test statistic by the square root of the sample size. Categorical data will be displayed as counts and percentages. Pearson and Spearman correlations will evaluate linear and non-linear relationships, respectively. Usability and secondary effectiveness will be analysed with the Wilcoxon signed-rank test, and improvement percentages will be reported for the experimental group. Changes over time will be compared using one-way Analysis of Variance (ANOVA) or Friedman’s ANOVA, with between-group differences assessed using independent t-tests or non-parametric tests based on data distribution. Statistical significance will be set initially at p<0.05, with confidence intervals reported. However, a Bonferroni adjustment will be used to control the increase in type I errors due to the conduct of multiple comparisons. Data from participants who discontinue or deviate from the intervention protocol will be retained in the study and analysed using an intention-to-treat approach, unless any of these participants request their data to be withdrawn, as outlined in the consent form.
Sample size
For a pilot RCT, Sim and Lewis32 recommend 20–75 participants per arm for medium to large effect sizes. To meet the minimum requirement, we initially planned to recruit 20 participants per group to gather preliminary data on the long-term usability and effect of the CUE1+ device while keeping recruitment manageable. This pilot RCT is the first two-arm study aimed at determining the appropriate sample size for a fully powered RCT. Our previous study was a 9-week feasibility single-arm study with 10 participants, which showed excellent compliance and no dropout rates. For this study, we decided to increase the sample size to 25 participants per group to allow for a 25% buffer for potential dropouts, due to the sham intervention. The results from this pilot study will inform the calculation of power and sample size for the subsequent fully powered RCT, helping to guide future research.
Study oversight
The principal investigators are VA, AN and CS. The research team will carry out the study on behalf of the investigators at QMUL. VA is the assessor and will remain blinded throughout the study together with the statistician who will perform the data analysis. KR, EC, KD, AZ, CQ, CB, TB and DG assist in participants’ identification and recruitment to achieve the recruitment target across two NHS sites. The Principal Investigators and research team will meet regularly to focus on recruitment and assessment targets. A trial steering committee will meet monthly to discuss and address recruitment, retention and review interim data analyses. The principal investigators will form a Patient and Public Involvement and Engagement (PPIE) group who will offer guidance and recommendations on future steps.
Patient and public involvement and engagement
Feedback from PwP and QMUL experts shaped the design of this study. PwP insights from the feasibility study and PPIE events with the East London Parkinson’s Group refined the research question, study design and materials like the protocol, consent forms and questionnaires. All PPIE events follow National Institute for Health and Care Research INCLUDE guidelines.33 PPIE members will receive comprehensive training. An advisory committee of 10 PPIE members will meet during the project (2-hour meeting) and post-project (3-hour face-to-face meeting). Meetings will include fair reimbursement for time spent and travel expenses for the last meeting that requires face-to-face attendance. During the meetings, we will seek feedback from PPIE members to address issues related to the study and guide future research and dissemination of results.
Ethics and dissemination
The study has received ethical approval from London-Dulwich Research Ethics Committee, London (reference: 23/PR/1526). Findings will be submitted for peer-reviewed publications. To share the study results, researchers will compile and analyse data into a Final Study Report, available to clinical and scientific communities. Findings will be published in journals, presented at conferences and disseminated through healthcare networks, including National Institute for Health and Care Excellence and associations such as the International Parkinson and Movement Disorder Society. Participants and their families will receive summaries, while NHS sites and Parkinson’s support groups will get targeted updates. Social media and news outlets will help spread the results further, with involvement from PPIE members for broader outreach.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors VA, AN and CS (along with PPIE members) were involved in the conception, design and writing and editing of the study protocol. VA, CS and AN were involved in the conception and editing of the protocol. VA, EB, KR, EC, KD, AZ, CQ, CB, TB, DG, AN and CS are involved in participants’ identification. VA is involved in participants’ recruitment and assessments. CS is responsible for randomisation and blinding procedures. All authors approved the final protocol. CS acted as guarantor.
Funding This work was supported by Knowledge Transfer Partnership (KTP) UK, 2021 to 2022, round 4, UKRI KTP (Innovate UK) and Charco Neurotech Ltd. We thank them for their support. AN is an external advisor to Charco Neurotech Ltd.
Disclaimer This article reflects only the author's view, and the Commission is not responsible for any use that may be made of the information it contains. The funders (KTP and Charco Neurotech Ltd) have no role or influence on the paper.
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.