Article Text

Abstract
Background Immunotherapies targeting the programmed death receptor-1/programmed death ligand-1(PD-1/PD-L1) checkpoint have a major impact on the treatment of both resectable and advanced non-small cell lung cancer (NSCLC). Additional blockade of the T-cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibition motif domain (TIGIT)-receptor may synergistically foster the immune-related response. Several trials are currently investigating the combination of neoadjuvant platinum-based chemotherapy and dual checkpoint inhibition prior to curative surgery. The investigator-initiated NeoTRACK trial (EU CT number: 2022-501322-38-00; ClinicalTrials.gov identifier: NCT05825625; IKF056) aims to evaluate the feasibility and safety of perioperative anti-PD-L1 (by atezolizumab) and anti-TIGIT (by tiragolumab) treatment in combination with chemotherapy in patients with early stage NSCLC.
Methods and analysis NeoTRACK is an open-label, single-arm, prospective, bicentric phase II trial. Patients with NSCLC in clinical stages II, IIIA and IIIB (only T3N2) will receive two cycles of standard platinum-based chemotherapy in combination with the anti-TIGIT antibody tiragolumab and the anti-PD-L1 antibody atezolizumab, followed by curative surgery. After surgery, patients without pathological complete response (pCR) will receive another two cycles of chemotherapy in combination with tiragolumab and atezolizumab, followed by tiragolumab/atezolizumab maintenance for up to 1 year (maximum 16 cycles). Patients with pCR will only receive dual immunotherapy. All patients will be followed-up for 30 months after the last study treatment. The clinical study will be aligned with a translational research programme to investigate treatment-naïve tumour tissues, surgical specimens and longitudinally collected blood samples. 35 patients are planned for enrolment. Patient recruitment started in August 2023, and treatment of the last patient is estimated to start 2.5 years thereafter.
Discussion The NeoTRACK trial aims to assess the feasibility and efficacy of combining tiragolumab and atezolizumab as both neoadjuvant and adjuvant therapies in patients with resectable NSCLC. The concept of treatment personalisation based on postoperative pCR is of great clinical interest.
Ethics and dissemination The trial obtained ethical and regulatory approval in Germany through the Clinical Trials Information System (CTIS, ID: 2022-501322-38-00) and the Paul Ehrlich Institute (PEI, competent authority for approval of clinical trials using medicinal products for human use in Germany, process number: PB00148) on 30 March 2023. A data safety and monitoring board will meet regularly to review ongoing treatment in terms of safety.
Study results will be published in peer-reviewed journals, presented at conferences and in the public registry of CTIS, following trial completion.
Trial registration number NCT05825625.
- Thoracic surgery
- ONCOLOGY
- Respiratory tract tumours
- CHEMOTHERAPY
- Immunology
- Lung Neoplasms
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
Novel multimodality treatment for locally advanced resectable lung cancer.
Sampling of treatment-naïve and treated patients using blood and tumour samples.
Longitudinal radiological follow-up and biosampling.
Open label non-randomised phase II trial.
Small sample size without placebo-controlled comparator arm.
Background
The introduction of checkpoint inhibition therapy has revolutionised lung cancer treatment within the last few years for stage IV non-small cell lung cancer (NSCLC) but also in patients with limited disease eligible for surgical resection. Recently, the efficacy of PD-1-targeted T-cell regulation by neoadjuvant immunotherapy was demonstrated in the phase III CheckMate 816 trial that included patients with resectable NSCLC. Induction treatment with nivolumab plus chemotherapy resulted in significantly longer event-free survival (EFS) and a higher proportion of patients with pathological complete response (pCR) as compared with chemotherapy alone. The trial also showed that the addition of nivolumab to neoadjuvant chemotherapy did not increase the incidence of adverse events (AEs) or compromise the feasibility of surgery.1
Anti-PD-1/anti PD-L1 therapies were also investigated in the postoperative setting showing positive benefit-risk for adjuvant atezolizumab2 and pembrolizumab.3
Recently, the results of the KEYNOTE-671 trial showed superior 3-year survival also for perioperative pembrolizumab (71%) as compared with placebo (64%; HR 0.72 (95% CI 0.56 to 0.93)).4 Perioperative durvalumab consistently showed meaningful benefit in EFS after 12 months however, overall survival data is immature.5
Another inhibitory receptor is the T-cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibition motif domain (TIGIT), which is expressed on activated T cells and natural killer (NK) cells. It has been shown to play an important role in modulating immune responses in the context of autoimmunity and cancer.6 7
TIGIT is co-expressed with other immune checkpoint receptors such as PD-1 on tumour-infiltrating lymphocytes in various tumours, including early-stage primary NSCLC.8 9 As TIGIT blockade was shown to act synergistically with PD-1/PD-L1 inhibition to enhance T-cell antitumour activity and also reactivate NK cell antitumour responses,10 we propose that combined administration of anti-TIGIT therapy with targeted PD-L1 therapy could reactivate antitumour immunity in NSCLC and provide clinical benefit to patients with anti-PD-1 resistant tumours.
The investigator-initiated NeoTRACK trial (NCT05825625; EU CT number: 2022-501322-38-00; IKF056) aims to assess the feasibility and efficacy of combining chemotherapy with tiragolumab and atezolizumab as perioperative treatment for surgical patients with NSCLC.
Methods
Trial design
This study is an open-label, single-arm, prospective, bicentric phase II trial. A total of 35 patients with stage II/IIIA and IIIB (only T3N2) NSCLC who are eligible for curative surgery will be enrolled. The objective of the study is to evaluate the feasibility and efficacy of tiragolumab and atezolizumab in combination with platinum-based chemotherapy as neoadjuvant and adjuvant therapy in patients with resectable NSCLC. Study participants will receive two cycles of standard platinum-based chemotherapy in combination with the anti-TIGIT antibody tiragolumab and the anti-PD-L1 antibody atezolizumab, followed by curative surgery. After surgery, patients without pCR will receive two additional cycles of chemotherapy in combination with tiragolumab/atezolizumab, followed by tiragolumab/atezolizumab maintenance for up to 1 year (maximum of 16 cycles). Patients with pCR will receive dual immunotherapy antibodies (IO-IO) only (without additional chemotherapy). All patients will be followed-up for 30 months after the last treatment.
Outcome parameters will be correlated to the accompanying translational analyses, which aim to understand and characterise the role of the tumour microenvironment in response to IO-IO therapies and to identify biomarkers that may be associated with response and resistance. Additionally, the safety of the therapeutic regimen will be described. Ethics committee approval was obtained (European Medicines Agency No: 2022-501322-38-00). All study-related investigations and enrolment of patients will only be conducted after written informed consent (online supplemental file 1) was obtained using the ethics committee-approved patient information and consent forms.
Supplemental material
Endpoints of this study
Primary endpoint
Major pathological response (MPR) after curative intent surgery: MPR is defined as ≤10% of residual viable tumour in lung and lymph nodes (evaluated by histopathological review).11
Secondary endpoints
pCR.
Radiological response by Response Evaluation Criteria in Solid Tumours (RECIST) V.1.1 criteria.
EFS: defined as the time from first dose of the study drug to any of the following events: disease progression precluding surgery, or by RECIST V.1.1, or death from any cause.
Overall survival (OS) is defined as time from treatment initiation (administration of first neoadjuvant cycle) to the date of death from any cause. A subject who has not died at the end of the trial will be censored at the last known date alive.
Rate of AEs and serious AEs rate. Safety assessments will include physical examinations including performance status (Eastern Cooperative Oncology Group (ECOG)), clinical laboratory profile and AEs.
All observed toxicities and side effects will be graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) V.5.0 for all patients and their relationship to the study treatment will be assessed and summarised. The rate of AEs and serious adverse events (SAEs) rate as well as the frequency of abnormal laboratory parameters will be determined.
Sample size calculation
This is an exploratory trial without testing of statistical hypotheses. No formal sample size calculation was conducted. Determination of sample size was based on the recruiting feasibility in this patient population.
A sample size of 35 with the assumption of an MPR rate of 36.9–57%1 12 results in an expected binomial proportion of 46.95%. The 95% Wilson-type CI for the MPR response rate is expected to have a width of 31.40% (the maximal width, which is observed if the MPR rate amounts to 50%, being 31.45%). The sample size calculation was conducted using the PASS V.16.03.
Assuming a screening failure rate of approximately 25% due to unexpected events, and detection of molecular alterations/mutations that at the time of this trial will presumably guide these patients to a different therapy scheme, it is planned to screen 46 patients for this study within a recruitment period of 30 months.
Patient selection/screening
Each patient must fulfil all inclusion criteria and none of the exclusion criteria for this study.
All patients (male and female, age: 18 years and older, ECOG performance status 0–1) with biopsy-proven NSCLC who are scheduled for surgical cancer therapy at the participating centres (Thoraxklinik Heidelberg, Universitätsklinikum Heidelberg and Ruhrlandklinik Essen, Universitätsmedizin Essen) will be screened for eligibility to participate in the NeoTRACK study.
The basic inclusion criterion is clinically confirmed stage II-IIIA and IIIB (only T3N2) NSCLC. The decision for curative surgical treatment must be made by an interdisciplinary tumour board with certification for thoracic malignancies.
At the same time, each potentially eligible patient will be re-evaluated by the investigator or authorised medical personnel. Only patients with adequate cardiopulmonary function according to current treatment guidelines will be included in the study.13 Main inclusion and exclusion criteria are listed in table 1 (refer to online supplemental file 2 for detailed in-/exclusion parameters).
Supplemental material
Main inclusion and exclusion criteria for NeoTRACK patients screening (refer to online supplemental file 2).
Patient recruitment and allocation
The official study start is listed as on 26 May 2023. Patient recruitment started in August 2023. A time frame of 2.5 years is estimated to allocate 35 participants. Assuming a duration of 1.5 years for treatment of the last patient included followed by an observation period of 2.5 years (follow-up), the total trial duration will be 6.5 years (first patient in to last patient last follow-up). Primary completion is estimated to be 1 November 2029. Study completion is estimated to be 1 February 2030.
Investigators will recruit patients directly during regular clinical consultation visits in the respective centre. All study-related investigations and enrolment of patients will only be performed after a written informed consent was collected using the ethics committee approved patient information and consent forms.
All patients who gave written informed consent will be captured online in the electronic case report form (eCRF) as screening patients to obtain a patient number which is used for pseudonymised identification throughout the study.
Provided that all other inclusion and exclusion criteria are met, the patient will be allocated and is allowed to start study treatment. The first treatment should be performed as soon as possible but not longer than three working days after allocation to the trial.
If one or more inclusion and/or exclusion criteria are not met or met, the patient will be marked as a screening failure in the eCRF.
Timeline
After successful enrolment, the treatment period will begin with the first IO-IO antibody application (day 1). The second application is scheduled for day 22. Curative surgery will be performed within 6 weeks after the last dosage of induction treatment. Preoperative direct imaging includes redo positron emission tomography (PET) scan for restaging and to assure continued technical resectability. Adjuvant therapy begins within the first 6 weeks postoperatively and depends on pathological response but is administered for a maximum of 16 cycles. After completion of adjuvant therapy, patients are followed for 30 months with assessments every 3 months (figure 1).

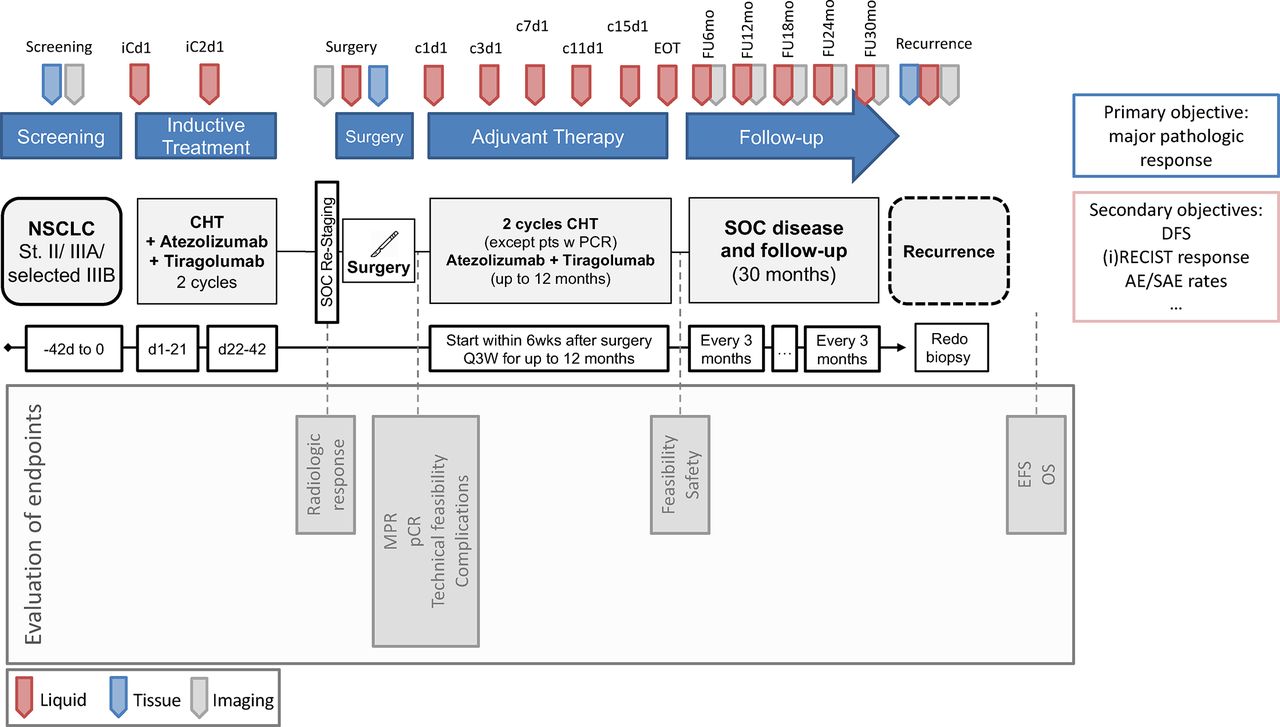{kind=link}
Study timeline. AE, adverse event; CHT, chemotherapy; DFS, disease free survival, EFS, event-free survival; EOT, end of treatment; MPR, major pathological response; NSCLC, non-small cell lung cancer; OS, overall survival; pCR, pathological complete response; RECIST, Response Evaluation Criteria in Solid Tumours; SAE, serious adverse event; SOC: standard of care.
Screening period
All eligible and consenting patients will have the following baseline/screening assessments performed prior to the start of study medication:
Tumour staging: contrast-enhanced CT scan of the chest for evaluation of local tumour situation; whole-body PET scan and an MRI scan of the brain to rule out distant metastases.
Histological confirmation of NSCLC is obtained by either transbronchial biopsy, transbronchial needle aspiration or transthoracic biopsy (ultrasound or CT-guided).
The mediastinum is assessed for enlarged and/or PET-positive lymph nodes using endobronchial ultrasound and transbronchial needle aspiration. Mediastinoscopy is performed if clinically indicated.
Clinical staging is performed according to the current eighth TNM (tumour-node-metastasis) classification.
The investigator will confirm the patient’s eligibility after reviewing all baseline scans and laboratory results.
Neoadjuvant chemoimmunotherapy
Patients will receive two cycles of neoadjuvant chemotherapy (investigator’s choice of platinum derivative) in combination with atezolizumab 1200 mg and tiragolumab 600 mg administered as intravenous infusion every 3 weeks. Chemotherapy is generally cisplatin or carboplatin combined with taxanes (paclitaxel, nab-paclitaxel, docetaxel), vinorelbine, gemcitabine or pemetrexed.
Surgery
Following neoadjuvant therapy, all study participants will undergo a new mandatory assessment by a multidisciplinary team prior to curative surgery. All patients are operated under general anaesthesia using an endobronchial double lumen tube. Surgery itself is not a specific trial procedure. The surgical approach (open vs thoracoscopic) will be individually determined and performed by the attending thoracic surgeons according to the patient’s risk factors. All patients undergo anatomical resection by lobectomy or bilobectomy with systematic mediastinal and hilar lymph node dissection. Intraoperative invasive lymph node dissection should follow the European Society of Thoracic Surgeons guidelines.14 Pathological examination includes local standard procedures for tumour staging and TNM classification. The resection margins (at least the bronchial stump) are examined intraoperatively for tumour remnants. The surgical specimen will undergo a further standardised examination according to the study protocol and international recommendations for specimen evaluation after neoadjuvant chemoimmunotherapy.15
Adjuvant therapy
All patients undergo a further multidisciplinary evaluation postoperatively. Depending on physical recovery and pathological response, adjuvant therapy is determined and initiated within the first six postoperative weeks.
No pathological complete response → two cycles of atezolizumab+tiragolumab+chemotherapy followed by atezolizumab+tiragolumab as maintenance therapy for up to 1 year (maximum of 16 cycles of adjuvant treatment) or until intolerable toxicity, disease progression or patient request, whichever occurs first.
Pathological complete response → atezolizumab+tiragolumab as maintenance therapy for up to 1 year (maximum of 16 cycles of adjuvant treatment) or until intolerable toxicity, disease progression or patient request, whichever occurs first.
Follow-up
After completion of adjuvant therapy, patients will undergo routine assessments every 3 months for up to 30 months, including chest and abdominal CT scans, pulmonary function tests and physical examinations. Patients with clinical or radiological evidence of tumour recurrence will undergo whole-body restaging, including a new biopsy for histology and other individualised assessments.
The investigators are experienced medical or surgical oncologists who treat patients in routine clinical practice. Therefore, the investigators will also further care for the patient after completion of the study treatment in the event of disease progression.
(Serious) Adverse events
The International Conference on Harmonisation Guideline for Good Clinical Practice defines an AE as ‘any untoward medical occurrence in a patient or trial subject who has received a pharmaceutical product that is not necessarily causally related to that treatment’.
An SAE is defined as any event that poses a significant risk to the patient. This includes incidents/events that are fatal or life-threatening, that could hypothetically result in death, that require hospitalisation, that result in permanent disability.
Hospitalisation for procedures required by the protocol or for the administration of study medication, hospitalisation for procedures planned before the start of the trial and elective hospitalisation are not considered SAEs.
Progression of the underlying malignancy is not required to be reported as an SAE or AE within this protocol (ie, a ‘tumor progression’ SAE or AE is not required to be reported), even if it meets a seriousness criterion (ie, fatal, requiring hospitalisation), unless the progression or symptoms of progression are causally related to the study drug.
Study discontinuation
The study will be discontinued if there are medical or ethical reasons not to continue, if medically unacceptable risks occur, if SAEs occur that are unexpected in type, duration or severity, or if known SAEs are expected to adversely affect the benefit-risk assessment.
Translational research
Tumour tissue and blood from all patients enrolled in the study will be collected by and stored for future biomarker analysis. For the accompanying translational research (TR), newly obtained core or excisional biopsies of a tumour at baseline and resections from surgery will be collected (according to standard procedures). This includes formalin-fixed, paraffin-embedded (FFPE) tissue blocks and, if the tumour bed can be identified in the fresh specimen, cryopreservation.
Further blood samples for TR are taken periodically during the administration of the first and second cycle of induction therapy, at the preoperative visit, on day 1 of cycle 1 and cycle 3 of adjuvant therapy and then every four cycles (approximately 12 weeks) during treatment and at the end of treatment. Blood samples will be taken before the study drug is administered. During follow-up, blood samples will be taken every 6–30 months as part of the TR project (figure 1).
In case of tumour recurrence before the end of the follow-up period, the last blood sample will be taken at the time of disease progression. If the recurrence occurs more than 30 months after the start of follow-up, the sample will be taken according to local practice. A tissue biopsy will be taken to confirm progression histologically and, if feasible and safe for the patient for the TR project. As part of ongoing longitudinal follow-up, these examinations should be repeated in the event of recurrence/progression.
Research summary of translational analyses
The effect of perioperative immunotherapy is influenced by a variety of parameters located within the tumour microenvironment, for example, tumourous PD-L1 and TIGIT expression as well as genetic alterations.16 The translational research programme therefore aims to characterise the peritumoural milieu by analysis of the patient blood and tumour-accompanying tissue.
Investigations on tissue samples
Tumour-adjacent cells will be analysed and quantified by immunohistochemistry and digital pathology. Whole-exome next-generation sequencing of tumour tissue will help to detect relevant genomic alterations as well as to define the tumour mutational load. To characterise the tumour microenvironment at the transcriptional level, messenger RNA (mRNA) from FFPE material will be used to create gene-expression profiles and analyse the activity of various immune-relevant signalling pathways, involved for instance in antigen presentation and cytokine activity.
Investigations on blood samples
Tumour-related and therapy-related immunological effects can be read from the peripheral blood by investigation of immune cells that circulate between the tumour microenvironment and the periphery. Peripheral mononuclear cells (PBMC) will be phenotypically characterised using flow cytometry. This may allow further analytical differentiation of T cells towards their state of activation (eg, T-cell exhaustion/reactivation/senescence). PD-L1 blockade promotes the growth of certain antigen-specific T-cell receptor (TCR) clones17 18 most likely originating from highly specific, antitumourigenic CD8+T cells. In order to detect therapy-induced changes in certain antigen-specific TCR clones T-cell DNA from PBMC samples will be extracted using TCR sequencing.
Cytokines represent messengers of the immune system playing a key role in the antitumour immune response. To address the systemic immune status at the functional level, we will create gene expression profiles to capture the activity of various immune-relevant signalling pathways that, for example, regulate antigen presentation. To capture the gene signature, mRNA is isolated from whole blood.
The data obtained through the translational analysis will be correlated with the clinical course. The aim is to elucidate immune-related mechanisms underlying the potentially synergistic, immune-stimulating effect of the combination of PD-L1 and TIGIT blockade in order to identify new biomarkers for predicting treatment response and for therapy control.
Statistical analysis
All data describing the study population, efficacy and safety will be analysed descriptively. Categorical data will be summarised with frequencies and continuous data with median, IQR or mean, SD, minimum and maximum. Survival endpoints (OS, event free survival (ESF), modified event-free survival (mEFS)) will be described by Kaplan-Meier estimates (including the numbers at risk for different time points) and median survival time.
The primary efficacy endpoint, MPR, is analysed in the primary analysis population (PAP) defined as all enrolled patients who underwent surgery. The MPR rate, defined as the number of patients with ≤10% of residual viable tumour in lung and lymph nodes as evaluated by pathology review, divided by the number of all patients with post-treatment tumour tissue available,11 is presented with 95% CI (Wilson-type). As a sensitivity analysis, the impact of missing values for the primary outcome will be explored using multiple imputation methods. Secondary endpoints pCR, technical resectability and perioperative complications are analysed in the PAP as well, whereas other secondary efficacy and safety endpoints are analysed in the as-treated population (ATP) defined as all enrolled patients who received at least one dose of any of the investigational (medicinal) products. The per-protocol population will be used for sensitivity analysis purpose and will comprise all patients of the ATP without major protocol violations such as wrong treatment received. Prognostic factors for MPR will be assessed via logistic regression, for which the area under the curve will be given with 95% CI. Factors of potential prognostic interest will be correlated to the outcomes MPR, pCR, EFS, mEFS and OS by means of calculating ORs or HRs by means of multivariable logistic or Cox regression models. Details of the statistical analyses are described in a statistical analysis plan finalised before database closure. All analyses will be performed in SAS V.9.4 (or higher).
Ethics and dissemination
The authors declare that they have followed the ethical standards of the responsible committees on human experimentation. The regulatory committees have declared that all regulatory requirements are in place to start with the NeoTRACK trial (EU CT number: 2022-501322-38-00). Approvals are available from the corresponding author on reasonable request. European Medicines Agency No: 2022-501322-38-00.
Written informed consent for scientific analysis of clinical details and/or associated medical data or study-related results is obtained from the participant.
The complete study protocol as well as all informed consent forms have been reviewed and approved by the sponsor and the ethical review committees. The relevance of scientific content and the compliance with the regulations for research with human subjects involved have been investigated.
Any subsequent modifications and amendments will also be reviewed by the investigators, the sponsor as well as by the ethical review institutions where applicable.
A Data Safety and Monitoring Board (DSMB) will meet regularly to review ongoing treatment in terms of safety.
All patient data is entered into the eCRF. All investigators and study nurses have been trained accordingly. Automatic validation during data entry checks the data for inconsistencies and enables confirmation or correction of this data by means of corresponding error messages. The validator must confirm that the data entered in the eCRF is complete and correct. After the database has been locked, the investigator receives a CD-ROM or paper copies of the patient data for archiving at the test centre. All patient-related data is recorded in pseudonymised form. Each patient is uniquely identified by a subject number, which is assigned upon enrolment in the study. The investigator must keep a patient identification log containing the full name and address of the subject and any other relevant personal data. Each participating site files the relevant documents (protocol, curriculum vitae (CVs), regulatory authorisations, etc) and trial-related correspondence in the site’s file, with an independent DSMB monitoring patient safety and performing a risk/benefit assessment.
Patient and public involvement statement
Patients and the public were not involved in any of the following issues:
Planning of the design and conduct of the study
Choice of outcome measures
Recruitment to the study
Discussion
The approval of immune checkpoint inhibitors has significantly changed the treatment options for patients with resectable NSCLC. The promising results of the CheckMate-816 phase-III trial have consequently led to the approval of neoadjuvant immunochemotherapies in surgical candidates harbouring tumours with high risk for recurrence.1
In addition, treatment concepts with neoadjuvant and adjuvant (perioperative) application of immunochemotherapy were recently developed. This strategy used in the KEYNOTE-671 trial demonstrated 3-year OS rates of 70–85% in patients with stage II to III NSCLC.18 But not all patients respond to anti-PD-1/PD-L1 treatment and it is worth considering whether there are options for patients who develop resistance.19
This highlights the need to develop new treatment algorithms, including the combination of different immunotherapies, to reduce the number of patients who develop recurrent or metastatic NSCLC.
Research indicates that TIGIT blockade combined with PD-1/PD-L1 inhibition enhances antitumour activity of T cells and also reactivates the antitumour response of NK cells.10 First clinical data from the CITYSCAPE trial20 showed promising results for dual immune checkpoint inhibition with tiragolumab and atezolizumab in patients with late-stage NSCLC. Dual checkpoint inhibitor therapy proved to be superior to atezolizumab monotherapy, especially in a subgroup of patients with high PD-L1 expression (>50%).20 21 In addition, the combination of atezolizumab with tiragolumab was well tolerated in the phase Ib part of the GO30103 study (NCT02794571) and the addition of tiragolumab did not alter the safety profile of atezolizumab.20 22 Of note, negative outcomes have been very recently published regarding combinatory tiragolumab/atezolizumab treatment in phase III NSCLC trials (SKYSCRAPER-01, KeyVibe).23 24 In brief, these trials aim to treat patients with stage IV NSCLC; some of them stratify towards PD-L1 expression levels. They therefore differ from the NeoTRACK population in terms of tumour stage, histology, therapeutic regimen.
Overall, improved multimodal treatment strategies are urgently needed to improve long-term survival after planned curative surgery. The safety profiles of anti-PD-L1 and anti-TIGIT antibodies generally compare favourably with classical platinum combination protocols, and there are no known specific adverse drug reactions (eg, wound healing problems) that would preclude the use of atezolizumab and tiragolumab in the (neo-)adjuvant setting.
We propose that combined administration of anti-TIGIT therapy with targeted PD-L1 therapy could reactivate antitumour immunity in NSCLC and provide clinical benefit to patients with anti-PD-1 resistant tumours. Furthermore, recent evidence questions the necessity and composition of additional adjuvant therapy in patients with complete pathological response after neoadjuvant therapy.18 We hypothesise that therapy-induced antitumourity in pCR patients is driven by immunotherapy, not chemotherapy. Patients with pCR tumours will therefore enter the adjuvant phase of dual-IO without receiving two additional cycles of adjuvant platinum-based chemotherapy, in contrast to patients with non-pCR tumours.
To date, PD-L1 serves as the only putative predictive biomarker in a combinatorial treatment approach, and our current knowledge of other biomarkers suitable for predicting response to dual immunotherapies is still in its infancy. This study therefore aims to identify additional biomarkers that may serve as parameters for response prediction. The spatial analysis of tumour samples that have undergone immunotherapy will significantly improve the understanding of the role of the microenvironment in the development of checkpoint inhibitor response but also help elucidate the mechanisms of resistance to these therapies. Perioperative systemic treatment concepts provide an excellent opportunity for a complete longitudinal analysis of the necessary tumour and blood samples to answer the above-mentioned research questions.
Ethics statements
Patient consent for publication
References
Footnotes
FB and FE contributed equally.
Contributors Conceptualisation: FE*; FB*; HW; MT; MEE. Methodology: FE; FB; HW; MT; MEE. Coordinating investigator: FE. Deputy coordinating investigator: FB. Mentoring investigators: HW; MT. Junior investigators: RMR; JS. Scientific coordination: PC. Sample collection and processing: MSchneider. Project management at coordinating investigator site: JC. Writing—creation of the original draft: RMR; FE; FB; JS. Writing—review and editing: RMR; JS.; LK; RG; MT; PC; ASt; MA; MSchneider; MSchuler; MW; ASc; SB; FD; BH; JC; MK; MEE; HW; FB; FE. Visualisation: RMR; FE; FB; JS. Guarantor: FE. All authors have read and agreed to the published version of the manuscript. *contributed equally.
Funding This is an investigator-initiated trial. The study design is based on the idea and concept of the investigators. Study sponsor is the Frankfurter Institut für Klinische Krebsforschung IKF GmbH, Frankfurt, Germany. Financial support and the provision of drugs for the conduct of the study is provided by the pharmaceutical company Roche Pharma AG, Grenzach-Wyhlen, Germany. The submitted publication of the NeoTRACK trial has been peer-reviewed by Sponsor before publication.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.