Article Text

Abstract
Introduction Multimodal anticancer therapies greatly damage the fertility of breast cancer patients, which raises urgent demand for fertility preservation. The standard options for fertility preservation are oocyte and embryo cryopreservation; both require controlled ovarian hyperstimulation (COH). However, there are safety concerns regarding breast cancer relapse due to the elevated serum estradiol levels during COH. Serum estradiol levels can be effectively decreased with the highly specific aromatase inhibitor letrozole. Letrozole is still uncommonly used during COH for fertility preservation, which has only been reported in a few studies, and the evidence of oocyte retrieval during ovarian stimulation and short-term safety from the perspective study is insufficient. As a result, this study will compare the efficacy of ovarian stimulation and the short-term safety of letrozole COH and non-letrozole COH protocols for preserving fertility in patients with breast cancer.
Methods and analysis This is an open-label, multicentre RCT being conducted in five Chinese reproductive medical centres. 64 eligible patients diagnosed with breast cancer will be randomly assigned (1:1) to the letrozole or non-letrozole group during their COH cycles. The primary outcome is the number of mature oocytes. The secondary outcomes are the number of high-quality embryos, incidence of ovarian hyperstimulation syndrome(OHSS) and recurrence rate of breast cancer.
Ethics and dissemination Ethical approval was obtained from the Ethics Review Committee of Guangdong Provincial People’s Hospital (KY-Q-2023-840-02). Written informed consent will be obtained from each participant. Findings will be disseminated to patients, clinicians and commissioning groups through peer-reviewed publications.
Trial registration number ChiCTR2300078625
- Breast tumours
- Randomized Controlled Trial
- Reproductive medicine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
This multicentre, randomised controlled clinical trial compares letrozole controlled ovarian hyperstimulation (COH) and non-letrozole COH protocols for fertility preservation in breast cancer patients.
Participants will be followed up to 2 years after fertility preservation, allowing the comparison of the long-term safety of the two protocols.
The trial will be an important ancillary study to investigate the safe COH protocol for fertility preservation in breast cancer patients.
Neither study participants nor investigators will be blinded, which could potentially introduce bias.
The sample size is determined based on the primary outcome, and the study has limited power concerning the assessment of the secondary outcomes.
Introduction
Breast cancer is one of the most common malignancies in women, with 2 308 897 new cases worldwide and 357 200 new cases in China in 2022.1 It affects women at least a decade earlier in China than in Europe and America,2 3 and the percentage of young patients under the age of 40 is greatly larger, accounting for approximately 15% of cases, whereas only 5% in the USA.4 Over the past several decades, long-term survival has been significantly improved due to the promotion of breast cancer screening and the development of comprehensive anticancer treatments. The 5-year disease-free survival rate exceeds 90% for patients in the early stage.3 However, multimodal gonadotoxic treatment has the potential to cause premature ovarian insufficiency, which reduces fertility significantly. Even worse, patients’ younger onset and postponed childbearing further exacerbate their reproductive issues. The optimal course of action is to preserve fertility beforehand, and the established standard options are oocyte and embryo cryopreservation, both of which require controlled ovarian hyperstimulation (COH).5 The COH protocol for breast cancer patients is still controversial.
A meta-analysis concluded that performing COH before or assisted reproductive technology (ART) following anticancer treatment in young women with breast cancer did not associate with detrimental prognostic effects on breast cancer recurrence, mortality or event-free survival.6 Despite encouraging results about the safety of oestrogen exposure during pregnancy and following ART in breast cancer survivors, conventional COH is associated with a substantial rise in oestradiol levels, which raises some safety concerns, particularly in hormone-responsive tumours.7–10 As a result, an alternate COH protocol involving the addition of a selective oestrogen receptor modulator or an aromatase inhibitor has been introduced to maintain low oestradiol levels.11 Letrozole is a third-generation, highly selective aromatase inhibitor that reduces oestrogen levels in the blood by inhibiting the conversion of testosterone to oestrogen. This alleviates the negative feedback on the hypothalamus and pituitary gland. The pituitary gland is essentially prompted to generate follicle-stimulating hormone (FSH), which facilitates follicle growth.12
For breast cancer, letrozole can be used as an endocrine therapy and medication to induce follicle growth. The administration of letrozole once per day in COH significantly reduced the serum oestradiol levels.8 9 13 Previous studies have revealed that the letrozole-antagonist COH (L-COH) enhanced ovarian responsiveness and increased the number of retrieved oocytes, mature oocytes and available embryos as compared with the non-letrozole COH (conventional antagonist COH, C-COH).14–16 Although the number of retrieved oocytes was similar in both groups, a retrospective investigation concluded that the oocyte maturation rates were lower in L-COH.17 However, a European prospective study found no difference in the number of retrieved oocytes and banked embryos between L-COH and C-COH.18
Two studies found there was no significant difference in the safety between traditional COH protocol and letrozole protocol among breast cancer patients.16 19 Previous prospective studies have focused on the number of retrieved oocytes or disease-free survival,18 19 whereas retrospective studies have indicated lower oocyte maturation rates in L-COH.17 Therefore, it is essential to investigate the efficacy of L-COH, with the number of mature oocytes as the primary outcome. Furthermore, compared with the Western population, the earlier onset age and higher proportion of young patients in China highlight the urgent need for prospective studies regarding fertility preservation in breast cancer.2–4,20 We intend to evaluate the efficacy and safety of the L-COH in breast cancer patients undergoing fertility preservation in this multicentre randomised controlled trial (RCT). The number of mature oocytes and high-quality embryos, as well as the incidence of OHSS, and the recurrence rate of breast cancer will be compared with that of the C-COH.
Methods and analysis
Study design
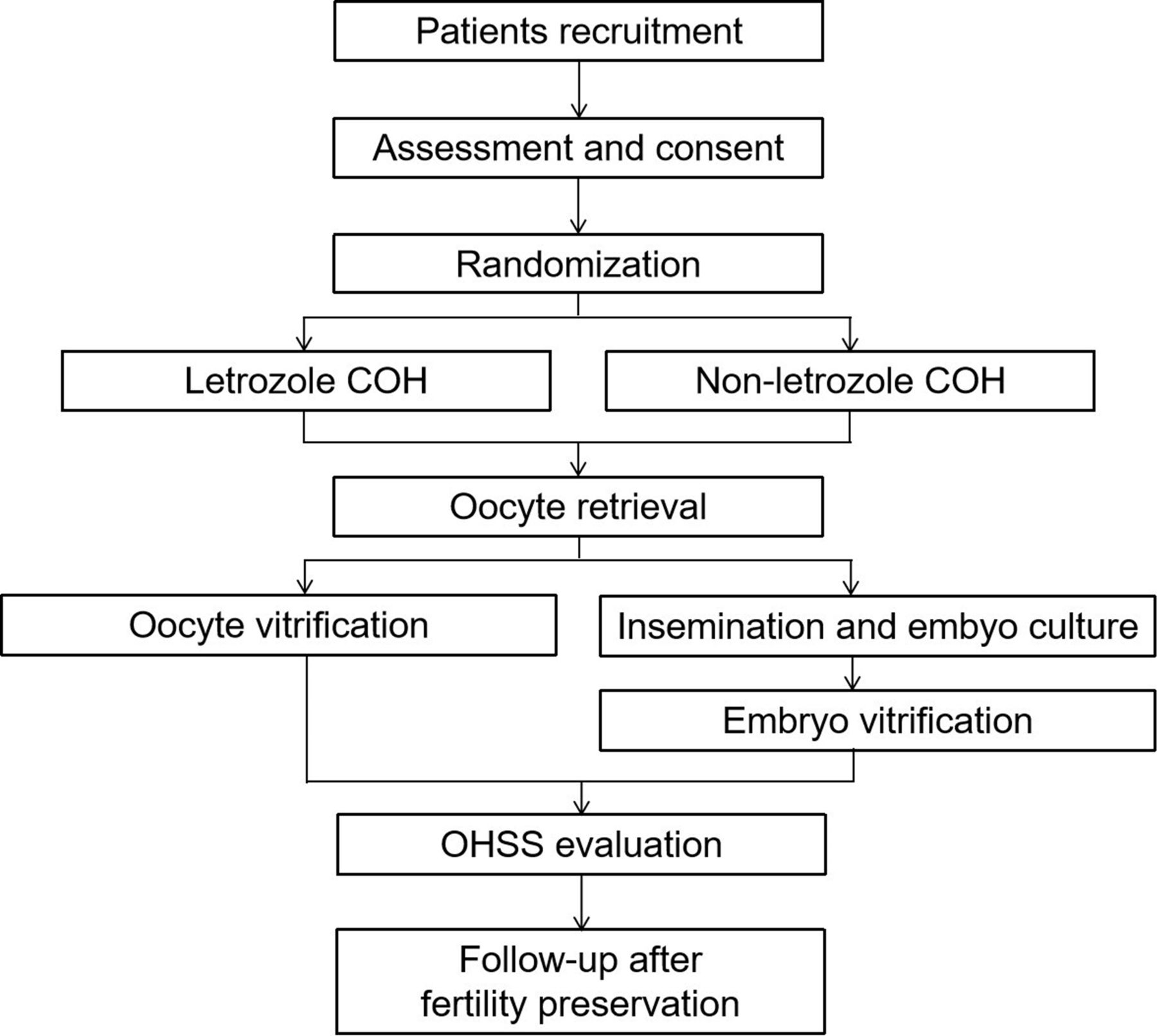
This multicentre RCT aims to evaluate the efficacy and safety of COH with or without letrozole for preserving fertility in breast cancer patients. In theirIn Vitro Fertilization(IVF) cycles, women will be randomised (1:1) to the L-COH or C-COH group. The number of mature oocytes and high-quality embryos, as well as the incidence of OHSS, will be monitored and analysed. This trial will adhere to the Standard Protocol Items: Interventional Trial Recommendations. The study period spans from December 2023 to December 2028. The enrollment, interventions and evaluation schedule are shown in figure 1, the study flow chart is displayed in figure 2 and the SPIRIT checklist is presented in (online supplemental file 1).21
Supplemental material

SPIRIT diagram for the schedule of enrollment, interventions and evaluation. Routine tests include the examination of blood routine, coagulation function, renal function, hepatic function and electrolytes. Tumour markers include CEA, CA153, CA125 and CA199. AMH: anti-müllerian hormone; COH: controlled ovarian hyperstimulation; FP: fertility preservation; OHSS: ovarian hyperstimulation syndrome.

{kind=link}
{kind=link}
Flow chart of subjects COH: controlled ovarian hyperstimulation; OHSS: ovarian hyperstimulation syndrome.
Study setting
Participants will be enrolled at four hospitals including Guangdong Provincial People’s Hospital, the Second Hospital of Hebei Medical University, Shenzhen Hengsheng Hospital,Nanfang Hospital of Southern Medical University and Women and Children’s Hospital, School of Medicine, Xiamen University. An independent data and safety monitoring board (DSMB) consisting of clinical, statistical and ethical experts will periodically oversee the trial’s progress and results.
Inclusion criteria
Eligible patients need to meet all of the following inclusion criteria:
Women with breast cancer.
Women aged ≤40 years.
Anti-müllerian hormone (AMH) level ≥1.1 ng/mL.
Exclusion criteria
Women will be excluded if they meet any of the following exclusion criteria:
Chromosome abnormality (except chromosomal polymorphism) of either partner.
A preimplantation genetic screeningis required.
Women with severe liver and kidney dysfunction or diabetes mellitus.
Recruitment
Fertility preservation will be recommended for all breast cancer patients who have fertility needs in the future. Eligible patients will be informed about the study’s specifics by the researchers, who will also allow them enough time to decide whether or not to take part in the trial. Women who agree to participate will be required to sign an informed consent form after the assessment. Signing informed consent involves several important steps to ensure that participants fully understand the study, its potential risks and benefits, their rights as participants and have an opportunity to ask questions. These steps include an introduction to the study, a discussion of key information and, finally, signing the consent form. The role of the research team is to support the participants in making an informed, voluntary decision without pressure or coercion. Women who choose not to take part in this experiment will receive routine clinical procedures. Each woman can only be enrolled once. Standardised case report forms are used to capture current medication status and previous medical history, including a detailed history of breast cancer. Patients’ names are substituted with their phonetic initials to protect their privacy. A transvaginal ultrasound scan and a physical examination (height, body weight, waistline, hipline and blood pressure) are carried out. Serum basal sex hormones including FSH, luteinising hormone (LH), estradiol (E2), progesterone (P), prolactin (PRL), testosterone (T) and anti-Müllerian hormone (AMH) will be measured. The results of routine tests will be recorded, such as blood routine, coagulation function, renal function, liver function and electrolyte examinations. In addition, tumour markers, including CEA, CA153, CA125 and CA199, will be measured before fertility preservation. Serum and follicular fluids will be collected and stored for future use in ancillary studies. The research protocol, V. 1.2, was completed on 20 November 2023.
Randomisation and blinding
The central randomisation system will be used to generate the random list in a 1:1 ratio between the two groups (L-COH and C-COH). The researchers can only obtain the assigned groups for the eligible patients by logging into the central randomisation system with unique identifications and passwords once a patient is recruited.
This study is an open-label trial. Participants, investigators, data collectors and analysts will not be blinded to the group assignment.
Interventions
Controlled ovarian hyperstimulation
Eligible patients are assigned at random to either the L-COH or C-COH group. The patients in the C-COH group will start receiving daily injections of gonadotropin (Gn) within 3 days of menstruation if the follicles are smaller than 10 mm and serum oestrogen and progesterone levels are at the basic levels. Gn dosage is determined by age, antral follicle count and basal sex hormone levels including FSH, LH and E2. Once the dominant follicle’s mean diameter reaches 12–14 mm, or after 5–6 days of Gn administration, a short-acting Gn-releasing hormone antagonist (GnRH-ant) will be administered daily until the trigger day. Serum sex hormones including FSH, LH, P and E2 and transvaginal ultrasonography are used to monitor follicle development. When at least two follicles reach 18 mm in mean diameter, human chorionic gonadotropin (HCG) or Gn-releasing hormone agonist will be injected for final oocyte triggering. Oocyte retrieval is performed 34–36 hours later.
In addition to receiving daily injections of Gn, patients in the L-COH group will also receive oral letrozole 2.5 or 5.0 mg until the trigger day. Patients weighing less than 40 kg will receive 2.5 mg daily, whereas others will receive 5 mg daily. The C-COH protocol will be followed for the remaining steps.
Oocyte vitrification and embryo vitrification
The maturity of the oocytes will be evaluated by the embryologists. Patients who are single or whose partners are unable to obtain sperm will have their mature eggs vitrified, and the remaining patients will have their oocytes inseminated. After keeping the fertilised embryos cultured for 3 –7 days, the embryologists will vitrify the cleavage embryos and blastocysts that are available. The number of mature oocytes, high-quality embryos and available embryos will be recorded.
Criteria for cycle cancellation
The COH cycle will be cancelled if any of the following criteria are satisfied.
No oocytes are retrieved.
Oocytes are not fertilised or fertilised abnormally.
No embryos are available.
Other private considerations.
OHSS evaluation
If the patient exhibits OHSS-related symptoms following oocyte retrieval, such as abdominal distension, nausea, vomiting, chest tightness, shortness of breath and oliguria, the OHSS grade will be evaluated, and the onset and duration of the symptoms will be documented. Patients with mild OHSS need close observation of their symptoms in the outpatient department. Patients with moderate OHSS require routine tests and ultrasound scan. Hospitalisation is usually necessary for patients with severe OHSS, and treatment plans involving particular interventions, such as intravenous infusion, albumin infusion, aspirin, and thoracic and abdominal drainage, must be documented. Each OHSS patient will receive ongoing care until their symptoms disappear.
Follow-up after fertility preservation
One year after fertility preservation, the medical records of the breast cancer patient will be monitored, taking note of the surgery time, surgery approach, pathology, TNM stage and clinical classification. The precise treatment regimen is documented if the patient needs adjuvant therapy, such as chemotherapy, radiotherapy, endocrine therapy or targeted therapy. Relapse will also be noted.
Two years after fertility preservation, we will monitor these individuals to see if they need targeted therapy or endocrine medication and to see if they relapse. We will also inquire about their pregnancy plans at the same time.
Outcomes
The primary outcome is the number of mature oocytes. The secondary outcomes include the number of high-quality embryos, incidence of OHSS and recurrence rate of breast cancer.
Mature oocytes are those in the MII stage. High-quality embryos are defined as cleavage embryos grade 2 or higher according to the Peter scoring system and blastocysts grade 4BB or higher according to the Gardner scoring system. The OHSS is classified as mild, moderate and severe categories according to the Golan criteria.
Statistical considerations
Sample size
According to a previously published study,18 the difference in the number of mature occytes between the two groups was 0.7. We assumed the overall SD was 3.8 with the cut-off value of non-inferiority of 4.0. Thus, the estimated sample size is 64 cases, with 32 cases in each group considering a power of 80%, an alpha error of 0.05 and a 10% follow-up drop-out rate.
Statistical analysis
Data analysis will be conducted using the SPSS (V.27.0; SPSS, USA). The trial will adhere to the intention-to-treat principle. Every test will have two tails, and a statistically significant result is defined as a p value less than 0.05. The Shapiro-Wilk test will be used to test whether the continuous variable is normally distributed. For normally distributed data, Student’s t-test will be used to examine the differences between the groups. However, for non-normally distributed data, the Mann-Whitney U test will be used. The mean±SD will be used for presenting continuous variables. Frequencies and percentages will be used for characterising categorical variables, and Fisher’s exact tests or x2 test will be used to examine the group differences.
Data collection and management
The data will mainly derive from medical records and telephone follow-up records following fertility preservation. An electronic data capture (EDC) system will be used to record and deposit the study data. A three-level data quality control will be performed to make sure the input data are accurate. The first step will be real-time logical and range checking built into the EDC. The second step will involve the validation of the original data by clinical researchers and remote data monitoring by EDC data managers, and the data entry staff will be responsible for the entry and correction of study data after receiving professional training. Careful data checks can help to detect more complex and uncommon errors. The third step will involve site visits by the DSMB to supervise the data. Errors found will be marked, and the staff responsible for data entry will receive notifications to confirm and fix the errors. Only authorised researchers will have access to consent forms, screening and identification records and other participant-identifiable data, which will all be stored in site files.
For those whose husbands were diagnosed with severe oligozoospermia, hypospermia and teratospermia, their oocyte outcomes but not embryo outcomes will be analysed. Women will be questioned about adverse events at each visit. Any adverse medical problems that occur during the study period are referred to as adverse events, regardless of whether or not they are related to the intervention. Every adverse event will be noted and sent to the DSMB. Furthermore, DSMB, which is independent of the sponsor and competing interests, will have the final decision on whether to terminate the trial.
Patient and public involvement
Patients and the public were not involved in the design, conduct, reporting or dissemination plans of this research.
Ethics and dissemination
This research has been approved by the Ethics Review Committee of Guangdong Provincial People's Hospital (KY-Q-2023-840-02). Each patient will be asked to sign a written informed consent prior to the interventions. The trial was registered on 14 December 2023, with the Chinese Clinical Trial Registry (ChiCTR2300078625). This research permits trial-related monitoring, audits and regulatory inspections. The results will be disseminated to patients, clinicians and commissioning groups through peer-reviewed publication.
Trial status
The trial is under recruitment and enrollment at the time of manuscript acceptance.
Ethics statements
Patient consent for publication
Acknowledgments
The authors thank all of the patients for their voluntary participation in this trial and the physicians at all study sites for referring subjects
References
Footnotes
YX, PL and WD contributed equally.
KW and YS contributed equally.
Contributors YX, PL, WD, KW and YS were involved in the study concept and design and in drafting of the manuscript. QF, JK, PS and KW contributed to the study design and critical revision of the manuscript. All authors contributed to the revision of the manuscript. All authors read and approved the final manuscript. YS is responsible for the overall content as guarantor.
Funding This research received the grant from China Preventive Medicine Association(840) and the Medical Research Fund of Guangdong Provincial People's Hospital (8200060682). Funders are not involved in the planning of the study, gathering, analyzing, and interpreting the data, or in preparing the manuscript.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.