Article Text

Abstract
Introduction Topical corticosteroids (TCS) of different potencies are the main treatment to control atopic dermatitis (AD). The Dutch guideline on AD for general practitioners (GPs) recommends a stepwise approach in which treatment steps are tailored to the severity of the disease, starting with the lowest possible potency of TCS. However, it remains unclear whether the recommended stepwise approach is most efficient. This randomised open-label controlled trial aims to determine whether a potent TCS is more effective than a low-potency TCS in the initial treatment of children with a moderate flare-up of AD in primary care. In the observational cohort, the overall aim is to determine the frequency, burden and determinants of flare-ups of AD during follow-up.
Methods and analysis The study is an observational cohort study with an embedded pragmatic randomised controlled, open-label trial. Eligible are patients diagnosed with AD (aged 12 weeks to 18 years) who visited the GP for AD or received repeated prescriptions for AD in the previous 12 months; follow-up of the cohort is 1 year. Children are enrolled in the trial if they have a flare-up of AD during follow-up in the cohort. Eligible children are randomised to the intervention group (with a potent TCS once daily) or to the GP guideline group (with a low potency TCS once daily). Primary outcome is the difference in average subjective disease severity over 24 weeks follow-up in the trial, measured with the patient-oriented eczema measure. As secondary outcome, the Eczema Area and Severity Index is measured.
Ethics and dissemination This study tests the hypothesis that immediate treatment with a potent TCS during a flare-up of AD leads to faster and more efficacious results as compared with starting with a TCS with low potency with less overall use of TCS. The study protocol is approved by the Medical Ethics Committee (MEC) of the Erasmus Medical Center Rotterdam, the Netherlands (MEC-2017–328). The results of the study will be published in international peer-reviewed journals and presented at national and international conferences.
Trial registration number NTR: 6679; Pre-results.
- Primary care
- paediatrics
- atopic dermatitis
- cohort
- randomised open-label controlled trial
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This is the first study to investigate the effectiveness of initial treatment with topical corticosteroids class I versus class III for long-term control of atopic dermatitis in children in primary care.
Pragmatic treatment strategy in real-life clinical practice.
Study is performed in general practice, where most children with eczema are treated.
Cohort study on determinants of flare-up and disease burden of atopic dermatitis in children.
This randomised open-label trial may be prone to observation bias.
Introduction
Atopic dermatitis (AD) or eczema is a chronic, highly pruritic inflammatory skin disease and one of the most common skin disorders in children.1 Eczema is in the top 10 of the most prevalent disorders in general practice in children aged up to 18 years.2 The prevalence of AD has increased over the past 30 years.3 It is estimated that 10%–20% of children and 1%–3% of adults in developed countries are affected by the disorder.4 In the Netherlands, cumulative incidence of AD at age 18 years is at least 24%.5 AD often starts in early infancy; about 45% of all cases begin within the first 6 months of life, 60% during the first year and 85% before 5 years of age.6 AD is associated with (later) occurrence of asthma and allergic rhinitis.5
The disorder results in significant morbidity and adversely affects quality of life (QoL). Factors that contribute to a poor QoL are fatigue, itch, activity restriction, depression and sleep deprivation.7 Approximately, 47%–60% of children with AD experience sleep disturbance,8 and children with AD and their parents can lose about 1–2 hours of sleep/night.9 Therefore, AD affects social functioning and psychological well-being, and has an even greater impact than diabetes on families of young patients.9 10 This includes direct and indirect financial costs, time spent on treatment, sleep deprivation (1–2 hours/night) and physician visits.
Since there is no definitive cure for eczema, suppressive treatment aims to control the disease. The majority of patients in general practice control their symptoms by application of emollients accompanied by symptomatic anti-inflammatory therapy consisting of topical corticosteroids (TCS).11 The Dutch guideline on AD for general practitioners (GPs) advocates a stepwise approach in which treatment steps are tailored to the severity of the disease, as determined using the Three Item Severity (TIS) score.12 The choice of potency of corticosteroids is determined by estimating the required effect. When AD is mild to moderate, a mild (class I) to moderate potent (class II) TCS is preferred, while potent (class III) TCS is used only when AD is severe. When treatment is insufficient, a higher class can be used. Due to safety concerns, the Dutch GP guideline recommends to use the lowest potency possible that will still be effective to treat the eczema. Potential local side effects of TCS are telangiectasia, atrophy, hypopigmentation and striae. However, the review of Siegfried et al showed no evidence of atrophy and supported the long-term safety of low and moderate TCS.13 Potential systemic effects of TCS may include suppression of the hypothalamic–pituitary–adrenal (HPA) axis, and osteoporosis, glaucoma, cataract and growth reduction. Nevertheless, osteoporosis and growth reduction are not reported in studies with long-term follow-up.13–15
The existing trials on the efficacy of TCS in children are often outdated and of inferior quality with only a short follow-up.16–18 However, they indicate that more potent TCS may result in faster and better disease control in AD; nevertheless, it is not clear which initial treatment strategy (ie, mild or potent TCS) is the best.12 19 During a flare-up, treatment with a potent TCS might lead to faster and better results as compared with starting with a mild TCS, with eventually less overall use of TCS. Besides improvements in disease control and patients’ satisfaction, this may also lead to fewer medical consultations and prescriptions, and may therefore be more cost effective. The present study focuses on these gaps and hopes to make an important contribution to knowledge regarding the use of TCS in children with AD treated in general practice.
Objectives
To determine whether initial treatment with a potent TCS is more effective than a mild TCS in the treatment of children with a moderate flare-up of AD in primary care in the short (ie, 1 week and 4 weeks of follow-up) and long-term (ie, 6 months follow-up) control of the disease.
In the observational cohort, the aim is to determine the frequency and determinants of flare-ups of AD. Furthermore, we will explore the burden of AD by measuring severity, medication use, healthcare use and QoL during 1-year follow-up.
Study design
The Rotterdam Eczema study is an observational cohort study with an embedded pragmatic randomised open-label superiority trial with two groups and a patient-reported primary outcome of long-term control. See flow chart in figure 1.

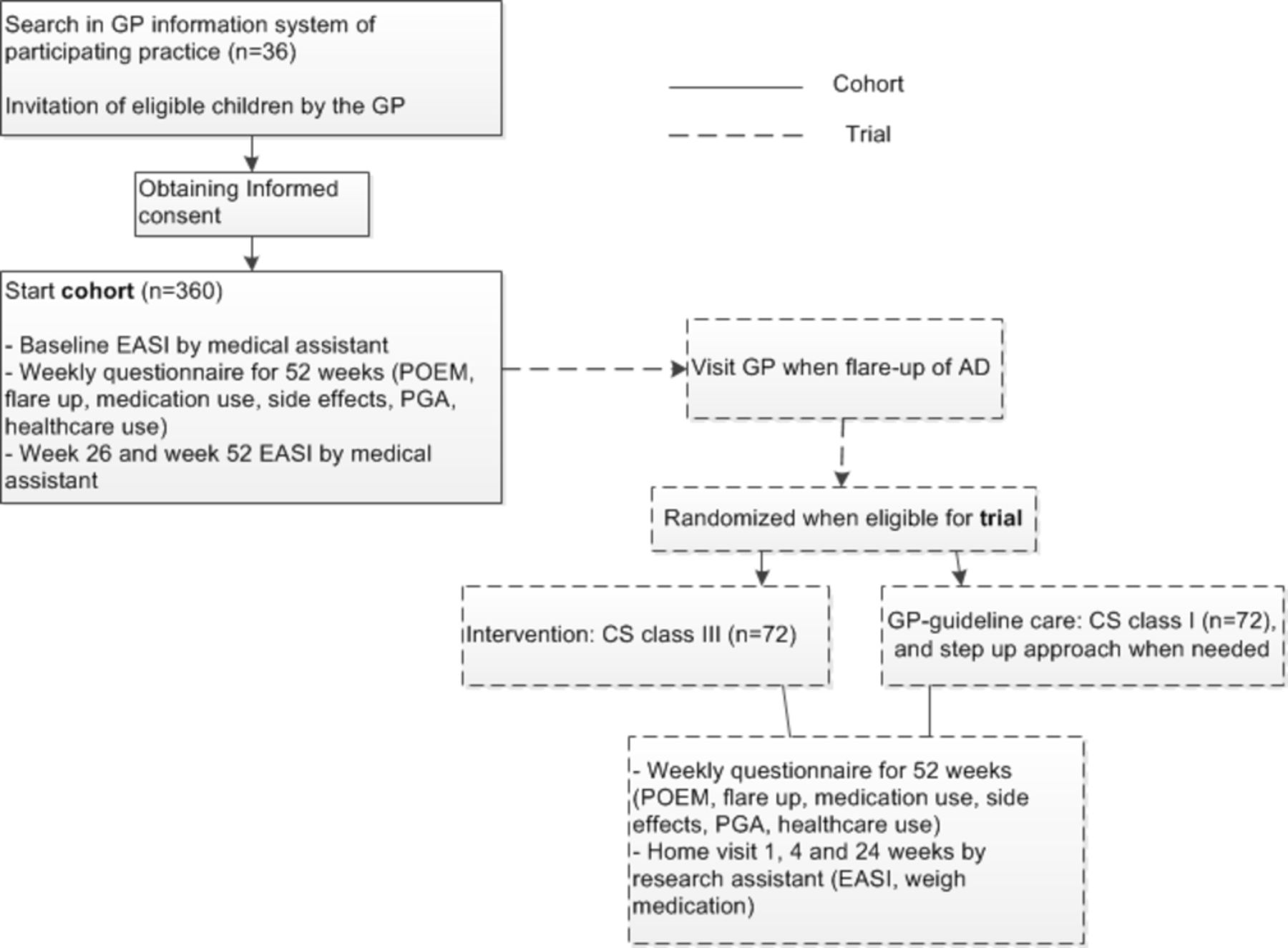{kind=link}
Flow chart of the study. AD, atopic dermatitis; EASI, Eczema Area and Severity Index; GP, general practitioner; PGA, patient global assessment; POEM, patient-oriented eczema measure; TCS, topical corticosteroid.
Methods; participants, interventions and outcomes
Healthcare system
The GP plays a key role in the Dutch healthcare system and (almost) everybody is registered with a GP practice. Diagnosis and treatment of eczema, also in children, are part of general practice. In case of diagnostic or treatment problems in children with eczema, referral to secondary care is available; however, referral to a dermatologist is not possible without the consent of a GP.
Study setting
Children will be recruited from general practices located in the Western part of the Netherlands. The GPs will perform a search in their information system to identify potentially eligible children.
Eligibility criteria
To be eligible to participate in the cohort study, a child must meet all of the following criteria: aged between 12 weeks and 18 years, diagnosis of eczema (International Classification of Primary Care (ICPC) code S87 and S88 or prescription of topical treatment for eczema) plus confirmation of the diagnosis by the GP, a consultation or repeated prescription in the previous 12 months and informed consent. This will be checked by the own GP and the researcher. Exclusion criteria for the cohort study are: as determined by the own GP. The own GP will check the list of selected children, the own GP is aware of potential problems limiting participation in a trial like family problems, for example, circumstances such as ongoing problems due to divorcing parents, relevant psychosocial problems, a seriously ill family member or if the child has serious comorbidities (eg, intellectual disability). Furthermore, exclusion criteria are: currently under treatment with a dermatologist, contraindications for the study medication, language barrier or no access to the internet (necessary to fill in weekly online questionnaire).
The inclusion criteria for the trial part of the study are: participation in the cohort, flare-up (ie, need to intensify topical treatment) from the child’s and/or parents’ point of view, a TIS score from ≥3 to <6. Exclusion criteria for the trial part are: use of TCS in the 2 weeks before inclusion in the trial, AD on eyelid(s), >50% of body affected by AD, other skin disorders hampering proper assessment of eczema, pregnancy and or breast feeding, or untreated skin infections based on clinical signs and symptoms (bacterial, viral, fungal or parasitic).
Interventions
Eligible children will participate in the trial part if they have a flare-up of AD. They will be randomised to either the intervention group or to the GP guideline group (control group). Those allocated to the intervention group will receive a potent TCS class III once daily to start with at each flare-up during the follow-up period of the trial. If children are aged >2 years, they will follow a predefined weaning-off scheme (ie, reduction of frequency) when their symptoms have improved. If children are aged <2 years, they will be reassessed by the GP after 1–2 weeks. When AD is improved but still needs treatment, children will be treated according to the Dutch GP guideline (ie, switch to a less potent TCS).
Children in the control group will receive care as stated in the Dutch GP guideline for all flare-ups during the trial period. First, they will start with a mild TCS class I once daily. When not improved within 1–2 weeks, a mild potent TCS class II once daily will be prescribed. When class II does not improve symptoms within 1–2 weeks, a potent TCS class III will be prescribed once daily. When symptoms do improve, children will follow a predefined weaning-off scheme.12
In the present study, hydrocortisone acetate cream 1%, and triamcinolone acetonide cream 0.1% will be used as class I and class II TCS, respectively, since this is a recommended preparation in the national guideline.12 For class III TCS, fluticasone propionate cream 0.05% will be used; this cream was chosen since it has a relatively short half-time as compared with the class III TCS recommended by the national guideline (ie, betamethasone).12 and is expected to limit potential side effects. Children will receive the prescription from their own GP and will obtain the prescribed medication from the child’s own pharmacy.
Besides the use of corticosteroids, all children (control and intervention group) will always be advised to use indifferent therapy with a standard emollient (ie, ‘cetomacrogol’). The advice is to use the emollients daily.
Patient and public involvement
The recommendations in the Dutch guideline are based on scientific research. It appears, however, that research to support recommendations is sometimes lacking or inadequate. The Dutch guideline states as a knowledge gap (‘lacunebak’) that it is not clear which treatment strategy, mild or potent TCS, is best when treating a flare-up of AD.12 This stated knowledge gap resulted in the research question of our study.
The outcome measures are based on the recommendations stated by the Harmonising Outcome Measures for Eczema (HOME)-initiative. During the development of the core outcome set and its measurement tools by this initiative, patients have been intensively involved.
The burden of the intervention is similar to the control group, both groups will use cream. Therefore, the burden of the intervention was not specifically discussed with patients.
Study participants will be informed about the results by a simplified summary send by email.
Outcomes
Primary outcome measure
The primary outcome will be changed in disease severity over 24 weeks follow-up in the trial, as measured by the average score of the patient-oriented eczema measure (POEM). POEM is a patient-reported outcome based on symptoms over the previous week, which can be self-completed by the child’s parent or the child. POEM is a validated questionnaire and has been recommended by the HOME initiative as the preferred instrument to capture patient-reported symptoms in eczema trials.20 The POEM score is collected weekly in the trial part over 24 weeks and, to measure long-term control, the difference between the two treatment groups in the average POEM scores will be measured over these 24 weeks.
Secondary outcome measures
The Eczema Area and Severity Index (EASI) will be used as objective measurement of the AD. The EASI has been identified by the HOME initiative as the core outcome measurement instrument to evaluate clinical signs of AD in all future trials investigating interventions for AD.21 The EASI is scored by a physician and rates both the intensity and extent of AD signs. The EASI will be used as secondary outcome in both the trial and the cohort study. The EASI score is collected at baseline, and at weeks 1, 4 and 24 of the trial, and at baseline, and at weeks 26 and 52 in the cohort study. See table 1 for an overview of observations made during the study.
Schedule of observations made during the study
Other secondary outcomes in the trial part include: (1) changes in disease severity after 1 week and 4 weeks using POEM, (2) QoL using the Infants’ Dermatitis Quality of Life Index (IDQOL) or Children’s Dermatology Life Quality Index (CDLQI) (depending on age), (3) medication compliance (determined as a POEM >8 and use of TCS during that week), (4) local side effects (painful application, telangiectasia, atrophy, hypopigmentation and striae), (5) time to recovery (ie, time till start weaning-off treatment), (6) frequency of flare-ups, (7) medication use (subjective and objective), (8) patient global assessment and investigator global assessment (both on a 6-point scale; clear, almost clear, mild, moderate, severe, very severe), (9) itch intensity score (the numeric rating scale-11 is ranging from 0, no itch to 10, worst itch imaginable) and (10) healthcare use (ie, telephone contact with GP, consultation at the general practice or referral to secondary care).
The QoL questionnaires are the IDQOL and CDLQI: both are validated and widely used in dermatology as a QoL scale for children aged ≤4 years and aged 4–16 years, respectively.22 23 The use of medication will be registered by weighing the tube of TCS after 1 week, 4 weeks and 24 weeks in the trial. Furthermore, in the weekly questionnaire, the children will register the number of days that TCS and neutral ointment were used in the previous week.
The aim of the observational cohort is to determine the frequency and determinants of flare-ups of AD after 1-year follow-up.
Secondary outcomes concerning the cohort includes disease severity at inclusion, and at 26 weeks and 52 weeks follow-up using POEM and EASI, QoL, frequency of flare-ups, medication use and healthcare use.
Sample size
In this study, we based our sample size calculation on the minimal clinically important difference (MCID) of the POEM in young children with eczema as determined by Gaunt et al.24 The MCID is the smallest change in an outcome measure that represents a clinically relevant outcome. A treatment effect of 3.0 POEM points was considered clinically relevant. We used a mean of 128.8 (SD of 5.9) as presented in trials with a similar population (ie, primary care patients) on baseline POEM characteristics.24
Taking these numbers into account, with a power of 80% and a=0.05 (two sided), we need 61 children per trial group. Assuming a drop-out rate of 15% during the study, 72 children per treatment arm are required.
Recruitment
Based on Dutch GP data and an expected patient participation of 50%, per participating GP, we expect to include seven children per year for the cohort, and four in the trial (based on flare-ups).2 Therefore, we expect to need about 36 participating GPs. First, we will collaborate with our academic network named PRIMEUR. The academic PRIMEUR network includes 13 centres with about 97 participating GPs and about 160 000 patients. If the inclusion is behind expectations, more GPs will be invited to participate in the study. Furthermore, if inclusion still remains behind expectations using the search protocol (eg, consultation, or repeated prescription in the previous 12 months), we will expand the search period from 12 to 24 months.
Children will be recruited from general practices. When a GP decides to participate, the GP will perform a search in their information system to identify all possible eligible children. Invitation letters to participate (in the name of the GP) will be sent to the parent/carer of the children.
Children are asked to respond using a response card or response email, irrespective of whether or not they are willing to participate. These responses are sent to the coordinating researcher of the department of General Practice, Erasmus Medical Center Rotterdam.
After receiving a positive response from the child, the research assistant will have telephone contact with the child (this can be with the parents if the child is aged ≤12 years or the children themselves). During this telephone contact, additional information on the study and study procedures will be explained. Based on the telephone contact, if the child is eligible and is still interested in participating, the patient information letter (PIF) will be sent by mail. Afterwards, if the child is willing to participate, the informed consent will be signed by the parents (if the child is aged ≤16) or the child (when aged ≥12 years) and sent to the department of general practice.
After receiving the signed PIF, the research assistant will contact the general practice of the child to inform them about the child’s participation. The GP practice will contact the parents of the child to arrange a consultation. During this first consult with the physician assistant, the baseline EASI will be obtained.
Assignment of interventions
When the AD flares up, the child (or the child’s parents) has to make an appointment by the GP. The definition of a flare-up is the need to intensify topical treatment from the patient’s and/or parents’ point of view
If the child has a flare-up of AD and is eligible for inclusion in the trial, the child will be randomly allocated to one of the two groups by the physician assistant of their own GP, using the data management system (research manager). The randomisation list will be computer generated and unknown to the investigators.
Children will be stratified by TIS score (ie, TIS score 3–5) to ensure equal distribution of the severity of AD between the intervention and control group. Random permuted blocks of two will be generated. The GP will prescribe the medication of randomisation.
Data collection and methods
Data collection of the patient-reported outcomes and the objective reported outcomes will be carried out using online case report forms. Children (or the child’s parents) will receive a reminder by email if questionnaires are not filled in after a standardised interval of 3 days. If questionnaires are not filled in at key time points, participants will receive a telephone reminder. Children will receive a small gift after completing the cohort or trial follow-up. The weekly questionnaires include a question on who completed the questionnaire, the child, the parent or together. We can, therefore, take into account by whom the questionnaire has been completed in our analysis. If patients are 16 years or older, they will complete the questionnaires themselves. Children under 16 are free in their choice to complete the questionnaire themselves or together with/by a parent. The physician assistants will received additional training in the pathophysiology and treatment of AD and in scoring the EASI.
Data management
Data will be handled confidentially and anonymously. A child’s identification code is used to link the data to the child; a unique code is randomly generated for each individual. The principal investigator safeguards the key to the code.
The software program research manager will be used for the online questionnaires and the children’s personal data, respectively.
Statistical methods
All analyses of the primary study parameters will be performed according to the intention-to-treat principle, that is, irrespective of compliance. Those who perform the analyses will be kept blind regarding which group has received what kind of treatment.
Secondary, a per-protocol analysis, excluding children in whom major violations of the protocol have occurred, will also be performed. Major protocol violations are: withdrawal from study or lost to follow-up, and medication compliance <75%.
In case major events occur during the study period that necessitate withdrawal from study, or lost to follow-up/drop-out for other reasons, weekly diary card data will be evaluated up to the week of such drop-out. However, children are requested to agree with further follow-up according to the study protocol (eg, weekly questionnaires). Medication compliance <75%, (also called non-compliance) is determined as a POEM >8 and no use of TCS during that week; the compliance is determined per week.
For the primary outcome, statistical comparison between the treatment groups will be done using analysis of covariance (ANCOVA) including the covariates: baseline symptom score, age and gender. Treatment effects will be tested two sided with a significance level of 5%.
For the main study parameter, it is essential that the weekly diaries are filled in adequately. However, in case of missing data, these will be imputed using multiple imputation; this is considered the most appropriate way of dealing with missing data.25 Missing values of the POEM will be imputed 10 times using the multivariate imputation by chained equations logarithm (R-Project). The imputation model included sex, age, type of medication used and frequency of application, and the outcome measure POEM.
Secondary outcomes of the trial, statistical comparison between the treatment groups for changes in disease severity, and QoL after 1 week and 4 weeks, will be performed using ANCOVA, including the covariates baseline symptom score, age and gender. The other secondary outcomes (ie, local side effects, systemic side effects, compliance, frequency of flare-ups, medication use and healthcare use) will be analysed with linear or logistic regression when appropriate. For the time to recovery, Cox regression analyses will be performed.
To explore differences in patient and disease characteristics in the two treatment arms, and to determine which factors are related to compliance to the two treatment strategies, backward logistic regression will be used.
Patient characteristics to be examined are: age, sex, age at presentation of AD, history of atopy (ie, asthma, allergic rhinitis, food allergy and anaphylaxis), use of other corticosteroid(ie, nasal, inhaled, oral) and QoL (IDQoL, CDLQI). Disease characteristics are disease severity (POEM and EASI), duration of AD, location of AD (ie, head and neck, upper limbs, lower limbs and trunk, all extracted from EASI) and previous medical care (ie, no previous treatment, GP only, GP and secondary care).
To explore which factors are related to compliance to the two treatment strategies, backward logistic regression will be used. The factors to be explored are treatment arm, age, sex, age at presentation of AD, disease severity (POEM and EASI), duration of AD, use of other corticosteroid (ie, nasal, inhaled, oral) and QoL (IDQoL, CDLQI).
The secondary outcomes for the cohort will be analysed with descriptive statistics (ie, disease severity, frequency of flare-ups, medication use, healthcare use, QoL). Analyses to determine what the determinants of flare-ups of AD are after 1-year follow-up will be performed with logistic regression analyses.
Ethics and dissemination
Amendments are changes made to the research protocol after a favourable opinion from the accredited MEC. All substantial amendments will be notified to the MEC and to the competent authority.
The results of the study will be published in international peer-reviewed journals and presented at (inter)national conferences. We aim to publish several peer-reviewed publications on the best treatment strategy in children with AD related to patient-oriented outcomes, healthcare consumption and side effects. The results of this study may be implemented into clinical practice and/or can be used for the next update of the Dutch guideline on AD for GPs.
Safety reporting
In accordance to section 10, subsection 4, of the Medical Research Involving Human Subjects Act (in Dutch: Wet Medisch-wetenschappelijk Onderzoek met Mensen), the sponsor will suspend the study if there is sufficient ground that continuation of the study will jeopardise subject health or safety. The sponsor will notify the accredited Medical research ethics committee (MREC) without undue delay of a temporary halt including the reason for such an action. The study will be suspended pending a further positive decision by the accredited MREC. The investigator will take care that all subjects are kept informed.
Monitoring
Due to the characteristics of this study it is not necessary to instal a data safety monitoring board. Nevertheless, the study will be monitored as described in the International Council for Harmonisation-Good Clinical Practice (ICH-GCP) guidelines (chapter 5.18). The department of general practice has developed a monitoring plan and monitoring checklist (based on the ICH-GCP guidelines) which will be used in this study. A senior researcher (project leader) will be designated as monitor. This senior researcher is not related to the current project and is part of another research discipline within the department. At various moments in the study (not known to the researcher in front) an appointment will be made with the researcher and project leader of the current project to monitor the study by making use of the checklist of the department of general practice.
Adverse events
All (serious) adverse events and suspected unexpected serious adverse reactions reported spontaneously by the subject or observed by the investigator or the staff will be recorded.
Discussion
This will be the first study to investigate the effectiveness of treatment with TCS class I versus class III on long-term control over 6 months follow-up of children in primary care with AD.
We chose an observational cohort design with an embedded pragmatic randomised open-label trial, as it might be difficult to randomise children in primary care at the moment they present with a flare-up. The observational cohort gives the opportunity to follow the course of AD in children in primary care with regard to the frequency and determinants of flare-ups and the burden of disease. As primary outcome, we chose a clinical outcome relevant to patients. In AD, the appearance of the skin does not always closely reflect the subjective symptoms, that is, when the latter causes a major impact on the child and family.9 10 Therefore, it was particularly important to design a trial with a validated participant-reported primary outcome.
The structure of regular home visits will probably artificially improve treatment adherence. It is generally known that treatment adherence is highest immediately after seeing a doctor.26 Both treatment arms will receive home visits, so this effect will be equally spread. The home visits will influence the generalisability of the results. However, it is crucial for several outcome measures to visit the patients. We kept the frequency of home visits to a minimum, balancing the unwanted effect of better treatment adherence and gathering important information on study outcomes.
A limitation of the study is the open-label design. Since participants know which treatment arm they will be assigned to, they cannot be blinded to the intervention. Also, because the GP must be able to adjust the treatment strategy or refer to the child if required, the GP cannot be blinded. The research assistants will also be aware of the medication use of the child, as they have to register and weigh the medication.
Given that our primary outcome is patient reported, the additional costs for blinding researchers to collect the objective data (ie, EASI) did not seem justifiable.
Concerns have been reported about the safety of TCS application in children with regard to incorrect application.27 Potential local side effects of TCS are painful application, telangiectasia, atrophy, hypopigmentation and striae. However, there is a lack of evidence from good quality research concerning these local side effects of TCS.28 Nevertheless, in a study on children with AD with a follow-up of 18 weeks, no difference was found in skin atrophy in children using class-III TCS for 3 days per week versus children using class I TCS.16 Potential systemic effects of TCS may include suppression of the HPA axis, and osteoporosis, glaucoma, cataract and growth reduction. Although there is lack of evidence about these potential systemic side effects, it is reported that topical TCS has little to no effect on the HPA axis, osteoporosis and growth reduction.12 14 15 29 In addition, the treatment scheme for the class III TCS is within the recommended dosage of the Dutch guideline on AD.12 Additionally, we want to include children of all ages with AD on most widespread areas. Therefore, we chose a strong topical corticosteroid with the most favourable profile, this is found in fluticasone propionate 0.05% or 0.005% since it has a relatively short half-life as compared with the class III TCS that is recommended by the Dutch guideline (ie, betamethasone).12
In this way, we aim to further reduce the already low risk of potential (systemic) side effects; therefore, we believe that it is safe to use a class III TCS according to the previously described treatment scheme.
Experience from dermatologists indicates that starting with a high-dose TCS leads to a faster and better result as compared with starting with a low-dose TCS. However, most children with AD are treated by a GP (only about 1% is referred to secondary care) and have a milder form of AD as compared with patients treated by a dermatologist.2 Whether the effects of initial treatment with a potent TCS as experienced in a specialist setting can be transferred to treatment in primary care is unknown. Since the present study will focus on these gaps, it will hopefully make an important contribution to knowledge with respect to the use of local corticosteroids in children with AD treated in general practice.
References
Footnotes
Contributors GE, AMB and PJEB conceived the idea and secured funding. Ethics applications were made by KFvH in collaboration with GE, AMB and PJEB. SGMAP and PJvdB provided intellectual input concerning study design and statistical analysis. All authors were involved in drafting or critically revising this work, and in final approval of the version to be published.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Ethics approval The study protocol is approved by the Medical Ethics Committee (MEC) of the Erasmus Medical Center Rotterdam, the Netherlands (MEC-2017–328).
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.