Article Text

Abstract
Introduction Spinal cord injury (SCI) has been linked to increased frequencies of sleep-related breathing disorders (SRBDs) (≤50% after paraplegia and ≤90% following tetraplegia). However, SRBDs have been under-recognised and undertreated among individuals with SCI. The OPTIMISE SCI (Outcomes Post Treatment: Impact on Motor Impairment of Sleep Efficiency in SCI) is an ongoing phase 3 clinical trial focused on the effects of the early use of continuous positive airway pressure (CPAP) therapy to treat individuals with moderate-to-severe SRBDs in the acute/subacute stage after SCI.
Methods and analysis A total of 44 participants with SCI who are newly diagnosed with moderate-to-severe SRBD are randomised into early CPAP therapy (initiated within the first 8 weeks postinjury) versus delayed CPAP therapy (initiated at 6 months postinjury). Participants with no/mild SRBDs are included in the control group (n=22). Primary outcome measures include neurological and functional recovery after SCI.
Ethics and dissemination The protocol for this randomised clinical trial (RCT) raised an interesting discussion with our research ethics board about delaying CPAP therapy by 3 months when a participant is diagnosed with moderate-to-severe SRBD. Given that the current standard of care does not include screening for SRBDs in individuals who are admitted for spinal cord rehabilitation, most individuals are screened for SRBDs during the chronic stage post-SCI, which represents a greater delay in the diagnosis and treatment of SRBDs in this population. Because the potential impact of the OPTIMISE SCI trial on the current standard of care outweighs the risk of delaying CPAP therapy by 3 months, this trial protocol was approved. The dissemination plan includes presentations at scientific meetings and publication of the results in a peer-reviewed scientific journal.
Trial registration number ClinicalTrials.gov (NCT05473689).
- Clinical Trial
- Neurological injury
- SLEEP MEDICINE
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
This randomised clinical trial includes a control group of participants with moderate-to-severe sleep-related breathing disorder (SRBD) who will undergo delayed continuous positive airway pressure (CPAP) therapy (based on the current standard of clinical practice) and a control group of participants with mild or no SRBD after acute spinal cord injury.
Adherence to CPAP therapy is anticipated to be a major challenge in this randomised clinical trial, even though the study protocol includes periodic communications with the research team to mitigate the participants’ concerns and difficulties.
The study participants will be recruited from a single tertiary spinal cord rehabilitation centre located in a large multicultural city under a publicly funded healthcare system.
Introduction
Background and rationale
Spinal cord injury (SCI) commonly causes substantial motor, sensory and autonomic deficits that impact many body systems, resulting in several secondary medical conditions (including sleep disorders).1 2 Sleep-related breathing disorders (SRBDs), including central, obstructive and/or mixed sleep apnoeas, are a common secondary medical condition after SCI, with a reported frequency of 67.9% in individuals living with chronic SCI.3 By considering a worldwide prevalence of traumatic SCI that reportedly varies from 50 to 906 per million inhabitants, 16–291 people are living with SRBDs after an SCI per million inhabitants.4 Notably, the results of a prior longitudinal study revealed that SRBDs are commonly present in the acute stage after SCI.5
In non-disabled individuals, untreated moderate-to-severe SRBDs have been linked with several psychosocial, neurocognitive and behavioural consequences that affect the individual’s quality of life, interpersonal relationships and occupational well-being.6–9 Restorative sleep is particularly important in individuals living with SCI given their motor, sensory and autonomic impairments, which make them more susceptible to a host of secondary health conditions, including SRBDs. For instance, individuals living with SCI are more prone to developing fatigue (54%–57%), depressive symptoms (29%–46%), anxiety (30%) and cognitive impairment (29%–60%).10–15 These secondary conditions have been associated with reduced social and community participation and have a negative impact on their employment and overall quality of life.16 Moreover, SRBDs can cause and/or aggravate fatigue, mood disorders and cognitive impairment after SCI, which can affect the individual’s participation in the initial rehabilitation during a critical period for their recovery.17
Continuous positive airway pressure (CPAP) therapy is the current treatment of choice for moderate-to-severe SRBDs. CPAP therapy has been shown to increase the calibre and decrease the collapsibility of the upper airways in the short and long term, respectively.18 Moreover, non-disabled individuals who are adherent to CPAP therapy often experience substantial symptomatic improvement (ie, reduced daytime sleepiness, improved quality of life and safer driving) and potentially reduced cardiometabolic risk.19 20 In people living with SCI, there is emerging evidence of the efficacy of CPAP therapy, but the differences between people with SCI and non-disabled individuals remain unclear. For instance, the results of a retrospective case-control study (that included 219 non-disabled patients and 25 patients with tetraplegia and SRBD) suggested that people with tetraplegia may require significantly lower CPAP to treat their SRBDs than non-disabled individuals.21
Also, SRBDs are often seen in individuals after other central nervous system (CNS) insults such as traumatic brain injury and stroke.22–24 Timely CPAP therapy has been shown to be effective in improving recovery after stroke. The results from four prior clinical trials revealed that CPAP therapy initiated between 3 weeks and 6 months after acute stroke, lasting 4 weeks to 2 years, significantly improved neurological and functional recovery among individuals who developed an SRBD following stroke.25–29
Given that SRBD remains a commonly and under-recognised medical condition after SCI, which typically results in delays in the diagnosis and treatment, we designed this randomised clinical trial to examine the clinically relevant clinical questions on whether:
Early use of CPAP therapy to treat individuals with moderate-to-severe SRBDs in the acute/subacute stage after SCI significantly ameliorates their neurological and functional recovery.
Early use of CPAP to treat moderate-to-severe SRBDs in individuals in the acute/subacute stage after SCI significantly reduces their cognitive impairment, fatigue, anxiety and mood symptoms and enhances their participation in inpatient rehabilitation.
Specific aims
The specific aims of this randomised clinical trial are to:
Evaluate the effectiveness of CPAP therapy in reducing motor and sensory impairment in individuals with SCI who developed moderate-to-severe SRBD.
Assess the effectiveness of CPAP therapy in mitigating disability in individuals with acute SCI who developed moderate-to-severe SRBD.
Determine whether CPAP therapy for moderate-to-severe SRBD is effective in improving cognition and mood, reducing fatigue and anxiety and ultimately, enhancing the participation of individuals with acute SCI undergoing rehabilitation.
Methods and analysis
Study design
OPTIMISE SCI (Outcomes Post Treatment: Impact on Motor Impairment of Sleep Efficiency in SCI) is a clinical trial that will compare 3 groups of individuals with acute or subacute, cervical or thoracic SCI (figure 1), as follows:
Early-CPAP therapy group where individuals diagnosed with moderate-to-severe SRBDs will start CPAP therapy within the first 8 weeks after SCI.
Delayed-CPAP therapy group where individuals diagnosed with moderate-to-severe SRBDs will start CPAP therapy 6 months after SCI, which would emulate the current standard of care.
Non-CPAP therapy group that will include only those who are diagnosed with no or mild SRBD.

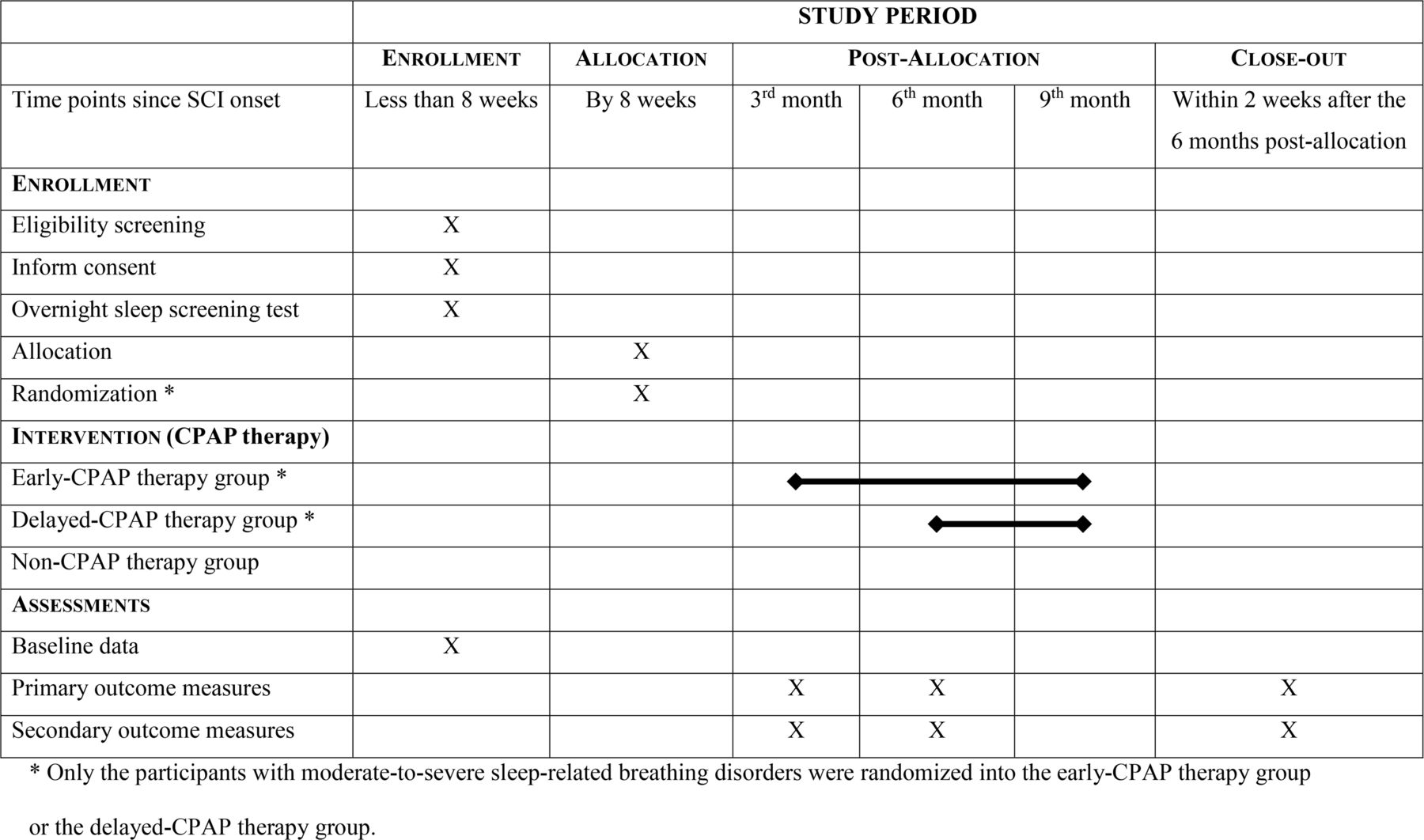{kind=link}
Study design including the schedule of enrolment, interventions and assessments. CPAP, continuous positive airway pressure; SCI, spinal cord injury; X: marks the protocol steps; horizontal thick lines: mark period of time when participants are on CPAP therapy
Notably, the protocol for this clinical trial was registered on the ClinicalTrials.gov website (NCT05473689) prior to initiating the recruitment of participants.
Patient and public involvement
This study design was established after obtaining the perspective of individuals living with SCI who underwent in-hospital unattended or home-based unattended sleep apnoea testing (unpublished data). Furthermore, the perceptions and limitations faced by the individuals living with SCI were studied in our multimethod clinical research that included home-based unattended sleep apnoea testing, followed by a 4-month trial of CPAP therapy and a qualitative study using a face-to-face interview with a structured questionnaire of open-ended questions (unpublished data; the study protocol is available at ClinicalTrials.gov ID: NCT04007380).
Moreover, the protocol for this randomised clinical trial was refined after input from a person with lived experience in delayed diagnosis of SRBD after SCI. This person supported the notion that individuals living with SCI could be screened for SRBDs within the first 8 weeks since the SCI onset. They also reinforced the use of in-hospital unattended or home-based unattended sleep apnoea testing for early diagnosis of SRBDs. Finally, they expressed no concerns about initiating CPAP therapy in the early phase after acute SCI, and, in fact, the inpatient team support was thought to facilitate the individual’s adaptation and adherence to CPAP therapy.
By the end of this study, every participant will receive a ‘white paper’ in English, where the main findings of the clinical trial will be summarised in lay language.
Recruitment and randomisation
This research project has been primarily carried out at the Lyndhurst Centre, which is a dedicated tertiary spinal cord rehabilitation hospital within the Toronto Rehabilitation Institute, University Health Network. De-identified data from the in-hospital unattended sleep apnoea tests will be transferred to Sunnybrook Health Sciences Centre for validation of the preliminary data analysis. Participants will be recruited from 15 August 2022 to 31 July 2026.
Screening for recruitment is performed by a research assistant from the Central Recruitment Service, which monitors all new admissions at the Lyndhurst Centre to identify potential candidates for clinical studies. Potentially eligible individuals who are identified by a research assistant from the Central Recruitment Service at Lyndhurst Centre and provide initial consent are subsequently contacted by the study research assistant. Those who agree to participate in the research project and fulfil inclusion and exclusion criteria are asked to sign the study consent form (online supplemental file 1).
Supplemental material
Those individuals who consented to participate in this study undergo an in-hospital unattended sleep apnoea test (using a Nox T3 device, Nox Medical, USA) within the first 8 weeks from the SCI onset. The first 44 participants with moderate-to-severe SRBD after SCI will be randomised into the ‘early-CPAP therapy group’ (n=22) and ‘delayed-CPAP therapy group’ (n=22) (figure 1). All participants with moderate-to-severe SRBD after SCI are randomised using an online randomisation system (Research Randomizer by the Social Psychology Network, Wesleyan University). A research assistant with a BSc degree has been in charge of the randomisation. The first 22 participants with no or mild SRBD will be enrolled into the ‘non-CPAP therapy group’ as a control group representing the natural history of neurological recovery after acute SCI without significant SRBD (figure 1).
Inclusion and exclusion criteria
This research project will include English-speaking adults (18 years of age or older) with acute (≤8 weeks after injury), cervical/thoracic (injury level at C2 to T12), complete or incomplete (ASIA Impairment Scale (AIS): A, B, C or D) SCI who were not being treated for SRBD prior to the spinal cord impairment onset. It will exclude individuals with a non-traumatic spinal cord disease at risk for neurological progression (eg, demyelinating spine diseases such as neuromyelitis optica and multiple sclerosis, and spinal cord malignant neoplasm), concomitant diseases of the CNS (eg, concomitant traumatic brain injury and stroke), preinjury chronic pain (eg, fibromyalgia, chronic pain syndrome, complex regional pain syndromes and postherpetic neuralgia) and other pre-existing diseases of the CNS (eg, previous traumatic brain injury, prior stroke, dementia, parkinsonism and multiple sclerosis), significant psychiatric disorders with a recent episode of exacerbation, neuromuscular diseases (eg, myasthenia gravis), current substance misuse, known history of primary hypersomnia (eg, narcolepsy or idiopathic hypersomnia) or secondary hypersomnia of any cause except for SRBDs (eg, hypothyroidism, moderate or severe iron deficiency anaemia (serum haemoglobin level below 110 g/L and/or significantly abnormal serum ferritin concentration), infections, depression, kidney failure, chronic fatigue syndrome, neurodegenerative diseases and myotonic dystrophy), epilepsy and vitamin B12 deficiency.30 Of note, hypersomnia is defined as prolonged sleep periods of more than 10 hours per night.
Diagnostic criteria of obstructive, central and mixed SRBD
A diagnostic polysomnogram in a sleep disorders clinic, which is the current standard of care, represents an access barrier for individuals with SCI due to limitations and challenges imposed by transportation, availability of attendant care and/or bowel and bladder management. The use of in-hospital unattended sleep apnoea testing (using a NOX T3 device) is a putatively practical, less costly, validated and reliable surrogate for the diagnosis of SRBDs.31–33 In a prior prospective study, Boulos et al demonstrated that a hospital-unattended sleep apnoea test is a feasible and practical diagnostic strategy for screening disabled people for SRBDs in their homes or hospitals without the need for an overnight stay in a specialised sleep clinic.33
Obstructive, central and mixed SRBDs are commonly defined according to diagnostic criteria from the American Academy of Sleep Medicine.34 35 For the purpose of this project, SRBD will be defined as an Apnoea-Hypopnoea Index (AHI) ≥5 events per hour of sleep. According to its severity, sleep apnoea is classified as mild if 5≤AHI<15 events, moderate if 15≤AHI≤30 events and severe if AHI>30 events.6
The recordings from the in-hospital unattended sleep apnoea testing are automatically analysed using the Noxturnal software as well as manually scored by a sleep physician according to the standards outlined in the American Academy of Sleep Medicine.35 As previously described, apnoeas were scored as ≥90% reduction in airflow for ≥10 s and hypopneas were scored as ≥30% reduction in airflow for ≥10 s with ≥4% oxygen desaturation.36 The sum of apnoeas and hypopneas per hour was calculated to generate an AHI. There is a board-certified sleep neurologist reviewing all in-hospital unattended sleep apnoea tests to ensure the clinical standards are met. Of note, the in-hospital unattended sleep apnoea test can differentiate central from obstructive SRBDs, as well as it can assess for hypoxia. Although no major changes regarding the participants’ medical condition or medications are anticipated during the period of 3 months between the randomisation and initiation of the intervention in the delayed-CPAP therapy group, the in-hospital unattended sleep apnoea testing will be repeated, if clinically indicated.
Participants’ baseline data
The following characteristics are collected from all participants:
Demographics: age, sex, ethnicity, level of education and socioeconomic status.
Injury characteristics: time since spinal cord impairment, degree of impairment as assessed using the International Standards for Neurological Classification of SCI (ISNCSCI) motor and sensory scores37 38 and degree of disability, as assessed using the Spinal Cord Independence Measure (SCIM).39
Pre-existing medical conditions: comorbidities as assessed using the Charlson Comorbidity Index, neck circumference, Body Mass Index, modified Mallampati classification and presence of retrognathia, macroglossia, tonsillar hypertrophy, elongated/enlarged uvula or high-arched/narrow hard palate.40–42 Of note, the modified Mallampati classification is based on the ‘relative position of the palate and base of the tongue inside the mouth—Class I: the entire oropharynx, including tonsils, pillars, soft palate and the tip of the uvula, is easily visible; Class II: the upper pole of the tonsils and uvula are visible; Class III: only part of the uvula and soft palate are visible and Class IV: only the hard palate and part of the soft palate are visible’.42
Current use of medications that may influence the study results, such as analgesics (in particular, opiates), gabapentinoids (eg, Neurontin and pregabalin), antispastic agents (eg, baclofen and tizanidine), anticholinergic agents (eg, oxybutynin, tolterodine and solifenacin), a synthetic cannabinoid, non-benzodiazepine hypnotic agents, benzodiazepines, melatonin and antidepressant drugs.
Furthermore, participants are screened for thyroid disease, anaemia, infection, renal dysfunction and vitamin B12 deficiency through laboratory screening tests that include: serum thyroid-stimulating hormone (TSH) level, serum ferritin level, complete blood count (including red blood cell count, haemoglobin, haematocrit, mean corpuscular volume, mean corpuscular haemoglobin, mean corpuscular haemoglobin concentration, white blood cell count and differential and platelet count), serum creatinine concentration, glomerular filtration rate, blood urea nitrogen and serum vitamin B12 concentration. If any screening laboratory test value is abnormal and clinically significant, the results are disclosed to the participant and initial treatment is recommended, or an immediate follow-up appointment is arranged with their primary care physician to ensure adequate care.
Outcome measures
Every participant with post-SCI, moderate-to-severe SRBD is evaluated with regards to the primary and secondary outcome measures at the time of recruitment (between 6 and 8 weeks after the SCI onset), as well as at 3 and 6 months after allocation.
The primary outcome measures include the degree of motor and sensory impairment using the ISNCSCI motor and sensory subscores, which is a comprehensive clinician-administered neurological examination for SCI. The ISNCSCI includes the motor subscores and the sensory subscores (ISNCSCI pinprick and light-touch sensory subscores). The entire examination includes testing and scoring 28 key points (dermatomes) for sensory subscores and 10 key paired points (myotomes) for motor subscores. The ISNCSCI score is the most accepted instrument for the assessment of the degree of impairment in the motor and sensory domains with excellent psychometric properties in the evaluation of the SCI population.37 38
The secondary outcome measures include:
The degree of disability using the SCIM, version III (SCIM), which is a clinician-administered disability scale developed to specifically evaluate the ability of individuals living with SCI to perform basic activities of daily living independently.39 The SCIM score varies from 0 to 100 and includes the following subscores: self-care (0–20), respiration and sphincter management (0–40) and mobility (0–40).39
The severity of fatigue is measured using the Fatigue Severity Scale (FSS), which is a self-reported (or administered by an interviewer) questionnaire. Most of the prior studies on the psychometrics of the FSS reported adequate to excellent validity and excellent reliability.43
The degree of depression and anxiety is measured using the Depression, Anxiety and Stress Scale-21 (DASS-21), which is a self-reported (or administered by an interviewer) questionnaire.44 The DASS-21 was reported as a valid screening tool for identifying, differentiating and assessing depression, anxiety and stress in patients with SCI.44
The degree of depression is measured using the Patient Health Questionnaire-9 (PHQ-9), which is a self-report screening questionnaire. The PHQ-9 was reported as a valid and reliable screening tool for identifying and assessing depression in individuals with SCI.45–47
The interference on the quality of sleep is measured using the Medical Outcomes Study Sleep Scale (MOS-SS), which is a self-reported scale that consists of six dimensions of sleep problems: sleep disturbance (ie, difficulty initiating or maintaining sleep), snoring, respiratory problems, sleep quantity, sleep adequacy and daytime somnolence.48 49
The degree of cognitive dysfunction is assessed using the Oral Trail Making Test.50 The time to complete each trail is measured, but a trail will be considered ‘incomplete’ if the individuals cannot complete it within 5 min.51
The degree of cognitive dysfunction is also assessed using a modified version of the Montreal Cognitive Assessment (MoCA test). The MoCA test has been shown to be a reliable, valid and responsive outcome measure for screening and reassessment of patients with cognitive impairment in stroke and other neurological diseases.52 The most significant limitation of the MoCA test in the SCI population is the fact that individuals living with tetraplegia are unable to complete tasks in the visuospatial/executive section, such as drawing and writing, due to their motor limitations. Given this, the original version 7.1 of the MoCA test was adapted for tetraplegics by replacing the figure copy and clock drawing with two visuospatial tests.
Intervention
After randomisation is complete, each participant will receive a device with the capability to be set up either as a CPAP or automatic positive airway pressure (APAP), which will be used to complete a 1–2 week auto-titrating trial. The auto-titrating CPAP/APAP device provides the least amount of pressure to the patient’s throat to ensure that their airway stays open during sleep. As the throat starts to collapse, the sensor tracks exactly how much pressure is needed to keep the patient breathing normally and signals the machine to raise or lower the air pressure accordingly. By measuring each breath, the auto-titration automatically adjusts the pressure both up and down.
Following completion of the 1–2 week auto-titrating trial, the participant will be provided with the results and informed that the device will be remotely set up for CPAP using the optimal pressure to eliminate episodes of moderate-to-severe SRBDs. In addition, a research assistant will communicate with the participants on a weekly or bi-weekly basis in order to verify if they have had problems using the CPAP device every night. At the end of each 3 months, the CPAP usage and adherence to the treatment will be remotely verified. Notably, the CPAP device (ie, ResMed AirSense 11, ResMed, San Diego, California, USA) used in this clinical trial is equipped with ResMed Airview, which is a cloud server for user adherence. A minimal adherence of 4 hours at night for at least 5 days a week will be considered adequate. According to our data from a prior 4-month single-arm trial of CPAP therapy to manage moderate-to-severe SRBDs, the above-described protocol, including weekly or bi-weekly communications with the participants and prompt support by a sleep technician, was sufficient to achieve adequate adherence to CPAP therapy (unpublished data; ClinicalTrials.gov ID: NCT04007380).
Notably, CPAP can also treat central apnoea in most individuals living with SCI. However, in the course of their clinical care, any participant will be considered for an additional in-laboratory titration study and be started on bi-level positive airway pressure or adaptive servo-ventilation if clinically indicated.
Blinding
The assessors and the statistician are blinded to group allocation. The research assistant will observe all assessments to prevent the participants from disclosing which trial group they are enrolled in.
Data analysis
Baseline data (including demographics, injury characteristics, pre-existing medical conditions, the results of the laboratory tests and the use of medications that could affect the study results) from the three study groups will be compared using the Kruskal-Wallis one-way analysis of variance (if non-parametric data) or the one-way Analysis of Variance (ANOVA; if parametric data) for continuous variables and Fisher’s exact test or χ2 test (for categorical variables). Data on outcome measures will be analysed using the intention-to-treat approach (which will count all participants, including the ‘crossover’ cases) and the per-protocol approach. Data on outcome measures from all three study groups will be compared at the three time points using repeated-measure ANOVA (for continuous variables) and Fisher’s exact test (for categorical variables). Missing data will be analysed using the imputation method.
Sample size
According to data published by Scivoletto et al and our preliminary unpublished data on motor and sensory recovery after SCI, an effect size for the ISNCSCI motor subscore was estimated to be 0.3732, and an effect size for the ISNCSCI sensory subscore was estimated to be 0.5110.53 With this, a sample size of 22 subjects in each group was estimated for data analysis using a two-factor repeated measures ANOVA, with a power of at least 80% and a significance of 5%, including a 20% loss to follow-up rate.54 Furthermore, in our prior prospective clinical study (unpublished data), a total of 19 individuals with chronic SCI was sufficient to show a statistically significant effect of CPAP therapy for 4 months on reducing their level of fatigue (p=0.0194; as assessed using the FSS) and the interference on the quality of sleep (p<0.0001; as assessed using the MOS-SS).
Feasibility analysis
Lyndhurst Centre admits approximately 300 patients with SCI every year, of whom 200 patients sustain a cervical or thoracic (injury level at C2 to T12), complete or incomplete (AIS: A–D) SCI.55 Of these, it is estimated that at least 124 patients (62%) will develop SRBDs after SCI, of whom 40 patients (32.1%) will be diagnosed with a moderate-to-severe SRBD every year.3 When considering a conservative screening-to-recruitment ratio of 2:1 for clinical studies, it is anticipated that the study will be completed within a 4-year period.56
Ethics and dissemination
This study involves human participants. This research protocol was approved by the Research Ethics Boards (REBs) from the University Health Network (CAPCR ID Number: 21–6214) and Sunnybrook Health Sciences Centre (Project Identification Number: 5623). Every participant is required to sign a consent form prior to being recruited into the study.
Ethical and safety considerations
The current standard of care includes the recommendation for CPAP therapy right after someone is diagnosed with moderate-to-severe SRBD. Nevertheless, the current standard of care in many other rehabilitation centres (including ours) does not include screening for SRBDs in individuals who are admitted for spinal cord rehabilitation. In fact, most individuals are screened for SRBDs during the chronic stage following SCI and after their discharge from the spinal cord rehabilitation centre, which represents a delay much greater than 3 months for the diagnosis and treatment of SRBDs among individuals living with SCI. Given that the current standard of care does not typically involve any sleep testing or treatment in most centres for this patient population, there is clinical equipoise, and it is ethically justifiable to delay treatment in the context of this important clinical trial.
Moreover, there are at least four publications on the results of randomised clinical trials that compared early-CPAP therapy versus delayed-CPAP therapy in patients with acute or subacute stroke, where no clinically adverse outcomes of the delayed CPAP therapy were reported. First, Ryan et al compared a group of 22 individuals with acute stroke (up to 3 weeks after stroke) who received CPAP therapy for 4 weeks with a group of 22 individuals with acute stroke who did not receive CPAP therapy.26 Although all participants underwent the same rehabilitation protocol, the former patient group had greater motor and functional improvement compared with the latter group, even though there were no significant improvements in neurocognitive outcomes, survival rate or risk for recurrent stroke, and there were no adverse events.26 Second, Khot et al compared 13 individuals with acute stroke who underwent CPAP therapy (for 17.0±9.5 days) with 17 individuals with acute stroke who underwent sham treatment (for 14.4±7.3 days).28 There were no statistically significant differences between the groups with regard to motor recovery, and no adverse events were reported for the non-CPAP group.28 Third, Gupta et al compared a group of 30 individuals with acute stroke who underwent CPAP therapy for 12 months with a group of 36 individuals with acute stroke who did not receive CPAP therapy for 12 months after diagnosis of moderate-to-severe SRBD at least 6 weeks after the first sign of stroke.27 While motor recovery was improved in the CPAP group compared with the non-CPAP group at 6 months to 1 year, there were no statistically significant differences between the two groups with regard to cognitive, sleepiness and blood pressure changes, as well as their rates of adverse events.27 Finally, Ren et al compared a group of 49 individuals with acute basal ganglia stroke who underwent CPAP therapy for 24 months with a group of 97 individuals with acute basal ganglia stroke who did not receive CPAP therapy for 24 months (‘control group’) after diagnosis of moderate-to-severe SRBD at least 6 weeks after the first sign of stroke.25 The authors reported that CPAP therapy was associated with reduced degrees of impairments and disability when compared with the group without CPAP therapy, even though no adverse events were reported for the non-CPAP group up to 2 years after acute stroke.25
Because the potential impact of the OPTIMISE SCI trial on the current standard of care outweighs the risk of delaying CPAP therapy by 3 months, this trial protocol was eventually approved by the REB of both institutions.
As a precautionary measure, a ‘crossover strategy’ was created for participants with clinically impactful SRBDs. When the clinical team identifies a patient with limited participation in the rehabilitation programme due to moderate-to-severe SRBD, they will be allowed to cross over to the ‘early’-CPAP therapy group if they were initially randomised to the ‘delayed’-CPAP therapy group. It is anticipated that the crossover strategy will be recommended for four or fewer participants during the course of this clinical trial. Regardless, these ‘crossover cases’ will be treated as additional participants with the aim of reaching 22 participants in each of the trial groups according to the above sample size estimation.
Dissemination plan
Once the study is completed, its results will be presented at scientific meetings, and, eventually, a manuscript with the final results will be submitted for publication in a peer-reviewed scientific journal. As mentioned above, every participant will eventually receive a ‘white paper’ in English, where the main findings of the clinical trial will be summarised in lay language.
Study data sharing
According to the governmental and institutional by-laws and regulations, the University Health Network (UHN) REB does not allow open access to the clinical data from any study. Should any external investigator be interested in obtaining our study data, they must request approval from the UHN REB prior to sharing the study data.
Ethics statements
Patient consent for publication
References
Footnotes
X @JulioCFurlan, @drmarkboulos
Contributors All the authors contributed to the study design. The first author wrote and revised the manuscript. The other authors reviewed and approved the final version of this manuscript. JCF is the guarantor.
Funding This research project has been fully funded by a grant from the Craig H. Neilsen Foundation (Grant Application ID number: 883905).
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, conduct, reporting or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.