Article Text

Abstract
Introduction Gait recovery remains one of the most determining factors in social participation for poststroke individuals, in whom ankle dorsiflexor function is closely related to gait speed. Focal muscle vibration has shown promising neurophysiological and clinical effects in neuromotor recovery. However, it remains to be determined whether tibialis anterior focal muscle vibration applied to the paretic limb could improve walking speed when implemented in early rehabilitation after stroke occurrence.
Methods and analysis This study describes a multicentric randomised controlled trial in which 70 participants will be randomly assigned in a 1:1 ratio to the tibialis anterior focal muscle vibration group or the sham group, in addition to their conventional rehabilitation. Participants will receive 100 Hz vibration/sham for 30 min, five times per week, for 8 weeks. The primary outcome will be gait speed, as assessed through a 10 m walking test and will be compared between groups at the end of the intervention. Secondary outcomes will include gait abilities, neuromuscular clinical evaluations and neurophysiological measures. Outcomes will be assessed at baseline and across five visits during and after the intervention, until 16 weeks of follow-up.
Ethics and dissemination Ethics approval was obtained from the French Ethics Committee ‘Protection des Personnes Nord Ouest III’ in 30 May 2023 (IDRCB: 2023-A00489-36). The results will be published in a peer-reviewed journal and presented at scientific conferences.
Trial registration number NCT05945212.
- Stroke
- Gait
- Lower Extremity
- NEUROPHYSIOLOGY
- REHABILITATION MEDICINE
- Randomized Controlled Trial
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This comprehensive investigation of gait recovery will encompass clinical, functional, mechanistic and neurophysiological outcomes.
Gait speed, a major determinant of quality of life in post stroke survivors, has been chosen as the main outcome
The 8-week intervention will consist of 30 min repeated focal muscle vibration sessions.
Due to the vibrating device conception, it was not possible to design a double-blind trial.
Introduction
Stroke remains the leading cause of acquired disability in adults, affecting approximately 12 million people worldwide each year.1 Although the ability to walk independently recovers in 65%–85% of survivors by 6 months after stroke, walking performance (ie, speed, endurance, pace, symmetry and balance) often remains impaired in chronic stroke individuals.2 3 According to the International Classification of Functioning (ICF), walking speed is correlated with overall physical and social outcomes.4 Therefore, current guidelines do recommend early poststroke rehabilitation, especially during the early subacute poststroke phase (ie, from 7 days to 3 months poststroke), when plasticity appears to be maximal.5 In this context, rehabilitation should target standing posture and walking, with the aim of improving autonomy. Noteworthy, while in healthy individuals walking speed is strongly related to ankle plantar flexors and hip flexors/extensors function, this differs in stroke survivors for whom walking speed is more closely related to the force production capacities of the paretic ankle dorsiflexors, regardless of the severity, location and extent of the lesion.6 In this context, early motor awakening (ie, regaining the ability to produce a voluntary muscle contraction) of the ankle dorsiflexors appears to be crucial as a first step towards strengthening these key muscles, improving walking performance and regaining autonomy. However, strategies specifically favouring muscular awakening still lack.
The use of focal muscle vibration (FMV) has recently shown interesting perspectives as an easy, safe, costless and painless strategy for neurological disorder rehabilitation, as reported in an umbrella review of systematic reviews.7 It relies on the fact that a single session of prolonged FMV can induce acute neural adaptations, suggesting its potential for neuroplasticity.8 9 Accordingly, its repeated application over weeks can induce long-term plasticity, leading to improvements in neuromuscular capacities for both upper and lower limbs in healthy participants.10 Specifically, studies have shown that the chronic application of FMV to relaxed muscles in healthy individuals can induce significant increases in force production capacities (ie, from 6.9% to 41%) of knee extensors,11 ankle plantar flexors12 and ankle dorsiflexors.13 Evidence of increased neural drive, which could have both spinal and supraspinal origins, was further associated with these increases in strength.11–14
For lower limb poststroke rehabilitation, and because of its ability to induce excitability modulations at different levels of the neuromuscular command loop, FMV may enable beneficial long-term neural adaptations.7 15 16 In a chronic poststroke population, a 6-week FMV programme targeting the tibialis anterior (TA) muscle and the two Achilles tendons, combined with weight-bearing activities in addition to conventional rehabilitation, was shown to significantly improve walking speed from an average of 0.38 m/s to 0.53 m/s.17 Similarly, a study focusing on a 4-week TA FMV programme in chronic ambulatory individuals, almost 2 years after stroke, showed an improvement in walking speed (ie, from 0.44 to 0.53 m/s), kinematic parameters during the swing phase, and in the average duration of activation of the TA muscle on the paretic side.18 Additionally, the randomised trial of Chen et al, in chronic poststroke individuals (ie, from 1 month to 2 years poststroke), showed greater improvements of the Functional Ambulatory Categories scale after 2 weeks of TA FMV in comparison with gastrocnemius FMV and control groups.19 Those improvements were further associated with spasticity reduction, preferentially in the TA FMV group.
While these studies in the chronic phase clearly highlight the interest of TA FMV, few studies have focused on its application in the subacute phase, even though this is theoretically the phase in which its application would be most relevant, given the window of cerebral plasticity.5 Yet, it remains to be determined whether TA FMV could improve motor impairment in poststroke survivors in their subacute phase.
Because TA FMV could be of real interest in the neuromotor recovery of the ankle dorsiflexors in the early subacute poststroke phase, we aim at evaluating its effect through an 8-week intervention by comparing neuromotor recovery between a group experiencing TA FMV versus a sham TA FMV group. Beyond impairments, it appears crucial to further assess the interest of FMVs in other dimensions of the ICF (eg, conceptualisation of individual’s level of functioning in daily life through a biopsychosocial prism).20 Thus, we further aim at evaluating locomotion abilities, as it is strongly related to quality of life outcomes.21 The primary outcome will, therefore, be gait speed, as assessed through a 10 m walking test (10MWT), and will be compared between groups at the end of the intervention. Secondary outcomes will include gait abilities, muscular testing and neurophysiological measurements. Our primary hypothesis is that gait speed will be clinically and significantly higher in the TA FMV group rather than in the sham-TA FMV group (at least +0.175 m/s). Our secondary hypothesis is that gait speed improvement will be supported by better clinical and neurophysiological motor improvements (ie, spasticity, ankle dorsiflexor strength, voluntary activation (VA), spinal excitability, quantitative and qualitative gait performance, level of autonomy and fatigue) in the TA FMV group during the programme, at the end and at follow-up.
Methods and analysis
Study setting
The NEUROVIB-AVC trial is a multicentric, randomised, controlled, single-blind superiority trial. Participants and evaluators will be blinded to group allocation, although the FMV operator will not be. The trial will involve two parallel groups. Details on the methodology are provided in online supplemental material following Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) reporting guidelines. The recruitment will be conducted in six rehabilitation departments, and all data collection assessments will be conducted at the Inter-university Laboratory of Human Movement Biology (LIBM, Saint-Etienne, France). The study will be proposed by treating physical and rehabilitation medicine physicians to all consecutive patients admitted to the participating rehabilitation departments, provided they meet the inclusion criteria and do not meet the non-inclusion criteria.
Supplemental material
Eligibility criteria
Eligible individuals must be male or female, aged between 18 and 80 years, experiencing their first hemispheric stroke (ischaemic or haemorrhagic) in the early subacute phase (ie, 14 days to 3 months). They must have a Medical Research Council (MRC) score of less than 4 on the ankle dorsiflexors of the affected side, no prior neurological condition likely to affect functional prognosis and be affiliated to the French social insurance system. Participants must also have received detailed information about the study and signed the informed consent form (online supplemental material).
Individuals not eligible for the trial will include those with a bilateral or infratentorial stroke, a history of orthopaedic ankle surgery interfering with measurements or who have received a botulinum toxin injection in the lower limb to be vibrated between stroke onset and enrolment. Contraindications to the use of the vibrating device will also exclude participants, such as those with superficial venous thrombosis within the past 3 months, deep venous thrombosis within the past 6 months, an elevated risk of thrombosis or those benefiting from any active device (eg, pacemaker, defibrillator, insulin pump, neurostimulator), presenting non-stabilised cardiac arrhythmias and/or epilepsy, or skin lesion close to the site for the placement of the vibrator. Additionally, individuals with significant psychiatric and/or cognitive impairments (ie, Mini-Mental State Evaluation <20/30) that may hinder comprehension of the protocol or adherence to the study procedures, as well as those under legal protection such as tutorship or curatorship, will not be eligible. Finally, participants already enrolled in other studies focusing on gait rehabilitation will not be included in the present study.
The sample size was calculated using RStudio software V.1.3.959 (RStudio, PBC, Boston, Massachusetts, USA) with 90% power of detecting a difference, 5% level of significance, assuming a difference in walking speed between the two groups (TA FMV or sham-TA FMV) of 0.175±0.21 m/s at the end of the 8-week FMV intervention.22–26 Considering a dropout rate of 10%, 35 participants are required in each group to detect a significant difference in walking speed.
Study design
All assessments will be conducted in the same laboratory (LIBM, Saint-Étienne, France). The evaluator, an experienced specialist in physical and rehabilitation medicine (HB), will be blinded to group allocation, ensuring that the assessment of all criteria remains unbiased. The primary outcome, gait speed, along with spasticity, ankle dorsiflexor strength, VA, spinal excitability, quantitative and qualitative gait performance, level of autonomy and fatigue, will be evaluated at baseline, and again at 4 and 8 weeks during the TA FMV (or sham-TA FMV) intervention (figure 1). Follow-up assessments will also be performed 4 and 8 weeks after the intervention ends. The timeline for participants is accurately described in table 1.

NEUROVIB-AVC study flow chart. FMV, focal muscle vibration; TA, tibialis anterior; NEUROVIB-AVC trial acronym.
Outcome measures throughout the study
Patient and public involvement
It was not appropriate to involve subacute poststroke patients or the public in the protocol methodology design.
Interventions
Participants will undergo an 8-week intervention consisting of either TA FMV or sham TA FMV, with 30 min sessions conducted 5 days per week (figure 1). The intervention will take place during hospitalisation in the rehabilitation department. Sessions will be supervised by trained nurses or physiotherapists who have completed a 1-hour dedicated training on device settings and session management. At the start of each session, the following standardised instruction will be provided to the participant: “A device that induces vibrations on your muscle will be attached to the front of your leg, like a watch. These vibrations are weak and you may not feel them due to your stroke. This is perfectly normal”. Session completion will be recorded for all the 40 sessions, along with any adverse events. If a session is incomplete, the duration and reason for interruption or non-performance will be documented. Detailed written instructions on session procedures will be distributed to all participating centres. The intervention will use the Vibramoov Physio device (Techno Concept, Manosque, France), configured to deliver vibration at a frequency of 100 Hz and an amplitude of 1 mm for 30 continuous min.16 For both sham and FMV groups, the wireless device will be placed on the muscle belly of the affected side, positioned at 80% of the distance between the lateral malleolus and the fibular head. During sessions, participants will remain seated comfortably in a chair with hips, knees and ankles flexed at 90° (figure 2).

{kind=link}
{kind=link}
Focal muscular vibration application: TA FMV or sham TA FMV intervention will be applied to the TA muscle belly, positioned at 80% of the distance between the lateral malleolus and the fibular head. The device will be secured to the skin using a dedicated elastic bandage. FMV, focal muscle vibration; ocal Muscle Vibration; TA, tibialis anterior.
All participants, irrespective of their group allocation, will receive conventional rehabilitation, as recommended by the French High Authority for Health. This rehabilitation focuses on preventing bedrest or immobility complications, avoiding capsular and musculotendinous retractions, improving trunk and limb mobility, strengthening muscles, training standing, balance and walking, as well as promoting independence in daily activities. Rehabilitation sessions will be tailored to each participant’s specific deficits, activity limitations and participation restrictions. These sessions will be conducted by a multidisciplinary team comprising physiotherapists, occupational therapists, speech therapists, physical activity therapists and psychologists. Participants will receive rehabilitation depending on their individual needs, with consistent access to comparable facilities and devices across all centres. A stratification will be done for each centre according to age (<50 years; ≥50 years) and NIHSS (National Institutes of Health Stroke Scale) scores (<16; ≥16). Individuals will be randomised into the two groups (1:1), using minimisation.
Outcomes
Primary outcome
For the 10MWT, participants will have to walk comfortably between two gates (WITTY·GATE, Microgate Srl, Bolzano, Italy) situated 10 m apart in a dedicated corridor with flat surface. The instruction will be to start walking 1 m before the first gate and to finish 1 m after the second one. As this outcome is recognised as a representative marker of the speed at which individuals will naturally walk for activities of daily living,27–29 participants will be allowed to use their walking aids and/or orthoses if needed.
Secondary outcomes
ICF-related impairments outcomes
Clinical assessments
The lower limb motor function will be assessed through muscular testing (MRC testing; scored out of 5 for each evaluated muscle, with a total score rated out of 60), the Fugl Meyer Assessment for lower extremity (FMA-LE; scored out of 34),30 31 and the level of spasticity of the lower limb using the Modified Ashworth Scale (scored out of 4 for each evaluated muscle, with a total score rated out of 20).32 33
Strength assessments
The maximal isometric ankle dorsiflexors voluntary force during contraction (MVC, in Nm) will be measured using a foot pedal fitted with a strain gauge torque metre (SMTR 500 Nm; Sensel Measurement, Vincennes, France). During the force measurement, the hip, knee and ankle angles will be 90°, 120° and 110°, respectively. The foot will be fixed to the pedal by two straps, and a harness will be attached at chest level in order to minimise upper body movements and ensure the safety of participants. During measurements, participants will be instructed to contract as strong as possible during 4 s and will be verbally encouraged by the evaluator. The maximum MVC value of the three recorded attempts will be retained. An interval of 1 min will be allowed between each MVC. Strength measurements will be performed on both legs (non-affected side first, then paretic leg), allowing for symmetry index calculation. During MVCs, surface electromyographic (EMG) signals will be collected continuously from electrodes (Meditrace 100, Covidien, Mansfield, Massachusetts, USA) positioned in bipolar configuration (interelectrode interval: 30 mm) on the TA. The EMG signals will be amplified (ML138, ADInstruments; gain=500), filtered (bandwidth: 5–500 Hz) and sampled at a frequency of 2000 Hz (PowerLab system, 16/30-ML880/P, ADInstruments, Bella Vista, Australia). Before placing the electrodes, the skin will be prepared (shaved, abraded and cleansed) to facilitate signal collection. The reference electrode will be placed on the lateral malleolus. The intensity of muscle electrical activity recorded by EMG during MVC will be calculated from the root mean square (RMS) value recorded over a period of 500 ms after the force has reached a plateau, with the maximum RMS value of the three recorded attempts being retained for analysis.
VA assessment
To approximate the voluntary neural drive that is directed to the TA muscle during contraction, the VA level (VA, in %) will be determined through a 4 s maximal ankle dorsiflexors MVC during which TA muscle electrical stimulation will be superimposed (100 Hz doublet; SIT). Another 100 Hz doublet stimulation will be delivered at rest (potentiated doublet, Dbpot), approximately 2 s after full relaxation. During measurements, participants will be instructed to contract as strong as possible and will be verbally encouraged by the evaluator. Two consecutive attempts will be performed, and the best one will be kept for VA calculation. The following formula will be used34:
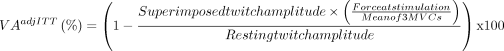
For muscular electrical stimulation, double-pulse 1 ms square-wave stimuli (maximal voltage: 300 V) will be delivered at 100 Hz over the TA muscle through a constant current stimulator (DS7A, Digitimer, Welwyn Garden City, Hertfordshire, UK) triggered by the Labchart software digital output (AD Instruments, Bella Vista, Australia). The cathode and anode electrodes (50×50 mm Durastick Plus electrode; DJO Global, Vista, California, USA) will be placed just below the tibial plateau, and approximately midway down the tibia.35 36 The stimulation intensity will be progressively increased by 10 mA until force plateaus. Data will be collected using LabChart software.
Spinal excitability assessment
For spinal excitability measurements, Soleus and TA EMG responses (M-wave and H-reflex, in mV) of the affected side will be recorded in response to tibial posterior electrical nerve stimulations in the popliteal fossa, before its fibular bifurcation,37 using a bipolar electrode with a Velcro strap allowing accurate positioning (Bar stimulating electrode, Digitimer, Welwyn Garden City, Hertfordshire, UK). The H-reflex is recorded when an electrical stimulus is applied to a peripheral nerve, causing action potentials in the Ia afferents of the muscle spindles, and recruiting in turn homonymous motor neurons. As such, the H-reflex is known as a tool to assess the effectiveness of Ia afferents in discharging motor neurons.38 The M-wave corresponds to the muscle compound action potential, its amplitude depending on sarcolemnal excitability. Stimulation intensity will be progressively increased by 2.5 mA steps until reaching a plateau for both Soleus and TA H-reflex amplitudes. Then, 10-mA steps will be used until reaching maximal M-wave amplitudes. One trial will be recorded for each stimulation intensity. For both TA and Soleus muscles, maximal peak-to-peak amplitude of the H-reflex will be expressed as a percentage of maximal M-wave amplitude (Hmax/Mmax).
ICF-related limitations outcomes
Gait abilities
In addition to the primary outcome, gait abilities will be evaluated through a 2 min walking test (2mWT, for long distance) using the distance covered (in m) in a 30 m straight flat corridor with cones at each end. We will also qualitatively evaluate walking performance through a three-dimensional (3D) Gait Analysis (only performed at week 8 and week 16). This consists of analysing gait through 12 cameras (Raptor-4 & Cortex software, Motion Analysis Corporation, Rohnert Park, USA) converging on 25 preset positioning reflective markers placed on participants (trunk, thighs, legs and feet), while participants walk on six force plates (90×90 cm, Model 9287C, Kistler, Winterthur, Suisse) and with wireless EMG electrodes (Trigno Wireless EMG System, Delsys, Natick, USA) recording bilaterally TA, gastrocnemius lateralis, vastus lateralis, rectus femoris and biceps femoris muscles activity. Participants will be instructed to walk at their own pace and along a 6 m designated path allowing kinematics, kinetics and EMG captures. Participants will also be allowed to use their walking aids and/or orthoses if needed.
Self-reported outcomes
The participant’s autonomy in walking activities will be assessed by the ABILOCO questionnaire (locomotion ability measure) which is a 13-item interview-based test focused on participant’s perceived difficulty in daily life.39 40 The responses are analysed using the Rasch model to convert the raw score into a linear measure, expressed in percentage of the range of measurement of locomotion ability (0=minimum ability, 100=maximum ability). In addition, the participants’ autonomy in the daily living activities will be assessed by the Barthel index, a summative 10-item score measuring different activities and the extent to which the participant is independent to perform them (scored out of 100, 0=minimal independence, 100=maximum independence), through a hetero evaluation.41 The participants’ perceived subjective fatigue will be further measured by the Functional Assessment of Chronic Illness Therapy questionnaire, a 13-item measure with 5-point Likert-type scales for each (scored out of 52, 0=maximal perceived fatigue, 52=no perceived fatigue).42 43
Assignment of interventions and blinding
Randomisation will take place at the end of the first evaluation (baseline before starting the vibratory/sham programme). Data will be centralised via an internet platform (Ennov Clinical software) where the investigator logs with his own identifiers. He proceeds with informatised randomisation after entering the participant data (inclusion number, monogram, age, validation of inclusion and exclusion criteria, centre number, and NIHSS scores). Participants and the evaluator (HB) will obviously not be informed of the protocol assignment and will remain blind to the group allocation. The blinded evaluator will feed data into the Ennov Clinical software in blinded datasheets so analysis can be performed without having access to information about the allocation.
Statistical analysis
The statistical analysis plan will be drafted blind to the data. The significance level for all statistical tests will be set at 0.05. Any deviations will be described and justified in the protocol and/or in the final report. This is an intention-to-treat study. All participants randomised in the protocol will be included in the statistical analyses in the rehabilitation group allocated to them. If missing data are observed, the analysis will be carried out on the intention-to-treat populations (with imputation of missing data) and using case-complete analysis (analysis of available data).
Descriptive analysis
The population included will be described overall and according to the rehabilitation group (TA FMV vs sham FMV) using the following statistics: quantitative variables (number of data available, mean, SD, median, first and third quartiles, minimum and maximum) and qualitative variables (absolute and relative frequencies expressed in %). The absence of imbalance between the groups will be checked on the initial variables and the known confounding factors. No tests will be performed, as differences should be evaluated in clinical terms.
Primary outcome analysis
We will compare the comfort gait speed (quantitative variable, in m/s) during the 10mWT after the vibration/sham programme (at week 8) between the two groups. It will, therefore, be presented in the form of mean, SD, median, first and third quartiles, minimum and maximum. The 95% CI of the means will also be calculated, as will the difference between the means and its 95% CI. The effect of TA FMV intervention will be estimated by comparing the gait speed at week 8 between the two groups. For this, a Student’s t-test will be performed if the variable follows a normal distribution. In the opposite case, a rank test will be used. The normality of the distribution will be verified by a Shapiro-Wilk test and a graphical representation (distribution histogram). The main analysis will be carried out without imputing missing data. If the primary endpoint is missing for some participants, a sensitivity analysis will be performed by imputing missing data, according to the principle of multiple imputation (in order to respect the intention-to-treat principle). In addition, a sensitivity analysis will also be performed on the primary endpoint by adjusting for randomisation strata. For this, an ANCOVA analysis will be used (covariance analysis). If an imbalance on one or more confounding factors appears, an adjusted analysis on this or these factors will be performed in order to check the consistency of the result. For this, an ANCOVA analysis will be considered. For this criterion (gait speed), a correlation will be established between the observed gain in walking speed (from baseline to week 8) and certain descriptive criteria such as age for each group. In addition, the delta in gait speed before/after rehabilitation for each group will be compared between the <50 and >50 age groups, using a Student’s t-test for non-matched data, if the distribution of the variable tested follows a normal distribution. In the opposite case, a rank test will be used. The normality of the distribution will be checked beforehand using a Shapiro-Wilk test.
Secondary outcomes analysis
The secondary outcomes are mostly quantitative endpoints repeated over time. Box-plots will be used to analyse the longitudinal data. In addition, differences in mean will be calculated, along with their 95% CIs. Correlation analyses will be carried out for exploratory purposes. Exploratory analyses will also be carried out on the different measurement times with the same ANCOVA analysis described above for each secondary outcome.
Discussion
Restrictions on social participation are primarily related to the stroke severity and sequelae limiting activities.44–46 In a daily activity and social role models for poststroke survivors, Faria-Fortini et al found that gait speed and ambulatory limitations were the two main predictive variables for social participation.47 It, therefore, appears that improving gait speed and its determinants would be an important factor in improving poststroke patient social participation. In this respect, early and focal interventions that target impairment in ankle dorsiflexors strength, a major determinant in gait speed, appear crucial.
FMV appears as a promising technique in several neurological impairments and contexts. Even if heterogeneous parameters seem to be widely applied,16 we decided to use a 100 Hz frequency and a 1 mm amplitude for FMV in this protocol, as both have demonstrated interesting neuromuscular effects on healthy participants and chronic stroke individuals, specifically for the ankle dorsiflexors.8 In healthy participants, after 8 weeks of TA FMV, MVC (+12%) and VA (+6%) were significantly improved, with interesting aftereffects measured 2 weeks after the end of such FMV application.13 The potential efficacy of TA FMV applied early in subacute poststroke individuals, as envisaged in the present trial (NEUROVIB-AVC), is further supported by the pilot study of Toscano et al, showing that a Rectus Femoris FMV protocol could statistically and clinically improve lower limb motor scores (FMA-LE score, with +10 points in the FMV group vs +3.5 in the sham group; and Leg Motricity Index, with +27 points in the FMV group vs +7.5 in the sham group) in acute poststroke individuals.48
We wished to implement this protocol in a ‘real-life’ approach, where most individuals are admitted to rehabilitation centres for several days after being discharged from stroke units in France. This allowed us to design the first multicentric trial including primary and secondary outcomes based on the ICF, which will comprehensively reflect the effect of 8 weeks TA FMV versus sham FMV in different dimensions of gait recovery after stroke. Each clinical outcome assessed (ie, gait speed, muscular clinical testing, spasticity) will be supported by a neurophysiological measurement (ie, 3D gait analysis, ergometry, H/M ratio) validated in the subacute phase poststroke population. We expect to measure the progressive effects of TA FMV during 8 weeks and their mid-term effect during the subacute phase. Neurophysiological approach we will use can objectively reflect muscular, spinal potential FMV effects that will be useful in understanding mechanisms that underpin stroke recovery and perspectives to foster it. Therefore, this trial will provide a more comprehensive and systematic protocol for further subsequent randomised controlled trial (RCT) with FMV interventions. This remains of particular importance as other studies using FMV are currently ongoing, for example, in upper limbs.49
This study protocol also has some limitations. Due to the FMV device conception, it was not possible to design a double-blind RCT, but in this study neither the participants nor the evaluator (HB) will know which group has been allocated. The source of data for the collection of cases will be geographically limited to one French region, which could not be totally representative of all French regions and population. However, the implementation of six centres will help in limiting extrapolability bias. This study also lacks long-term (>6 months) follow-up and evaluation, and the long-term effects of the intervention are unknown. The heterogeneity of brain lesions will necessarily lead to different participant phenotypes, but we will be able to identify whether or not poststroke survivors respond to FMV, in the ‘real-life’ clinical context.
Throughout this study, we believe that this safe and non-invasive bottom-up technique may play an important role in lower limb neuromotor recovery,7 48 but this remains to be demonstrated before implementing FMV as an adjuvant/complementary therapy in poststroke gait rehabilitation.
Ethics and dissemination
Data management and monitoring
Data management will adhere to European General Data Protection Regulations, with all data stored using participant numbers recorded in the secured Ennov Clinical software. The data will be available on reasonable request. French regulations require a paper trail for consent forms, completed measurements and adverse events, which will be stored in a locked facility. Project progress and data updates will be maintained on the Clinical Trial Register, upon University Hospital Center of Saint-Etienne responsibility (the study promotor). The data will first be collected in case record form (CRF) as and when the study visits take place. The data will then be entered, under the investigator’s responsibility, by a member of his or her team on an electronic CRF (e-CRF) (above-mentioned Ennov Clinical software). All adverse events that occur during the intervention and follow-up will be recorded on the eCRF and will be evaluated for relevance to the intervention. The occurrence rate of the adverse event (number of occurred participants divided by all participants) will be calculated. The database will be frozen before statistical analysis. Data that will support our findings will be available upon reasonable request.
Ethics approval was obtained from the French Ethics Committee ‘Protection des Personnes Nord Ouest III’ (IDRCB: 2023-A00489-36) in 30 May 2023 (Trial registration number: NCT05945212) for all the participating centres. In accordance with the methods outlined, all participants will undergo medical evaluation and monitoring throughout the study.
The SPIRIT guidelines were used to report the study protocol.50 The results will be published in a peer-reviewed journal and presented at scientific conferences with relevant clinical and scientific professionals. The number of articles published will depend on the limits imposed by the number of words and the scope of the scientific journals for which the data will be most important and adequate. Given the complexity of the data obtained over the 16 weeks of testing, we believe that a single article may not adequately present and discuss all findings. However, an overview booklet will be prepared for all study participants and professionals enrolled in the protocol.
Trial status
The trial is ongoing and recruiting participants. This study protocol is version 2 made on 20 July 2023. Recruitment started in March 2024 and is expected to be completed in January 2027.
Ethics statements
Patient consent for publication
Acknowledgments
This study is supported by the University Hospital Center of Saint-Etienne, and affiliated rehabilitation centres (University Hospital Center of Lyon, Hospital Center of Rive de Gier, Hospital Center of Chambon-Feugerolles, Hospital Center of Roanne and 'Le Clos Champirol' rehabilitation centre). We thank all the professionals who supported this research.
References
Footnotes
Contributors HB, BF, DR and TL conceived the study, designed it and prepared the protocol. Original draft was written by HB. VM, NE, MV, CA, BF, DR and TL revised the manuscript. TL, BF and DR supervised the final manuscript edition. The Clinical Research Unit of the University Hospital of Saint-Etienne provided statistical expertise in clinical trial design. BF and DR are the supervisors of each randomisation. TL is the scientific supervisor. HB is the blind evaluator all along the study. CA, VM, NE and MV will provide technical support for experiments. TL is responsible for the overall content as guarantor.
Funding This study was supported by the French Ministry of Health. The University Hospital of Saint-Étienne has promoted this study.
Disclaimer This funding source had no role in the design of this study and will not have any role during its execution, analyses, interpretation of the data, or decision to submit results.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; peer reviewed for ethical and funding approval prior to submission.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.