Article Text

Abstract
Introduction Chronic non-cancer pain presents a global health problem, with a significant increase in opioid prescriptions over recent decades. However, opioid therapy poses risks of adverse events, overdose and non-medical use. As a result, many patients seek to discontinue or reduce their opioid intake. Strategies for opioid tapering often lack efficacy, prompting the investigation of novel approaches like open-label placebo (OLP), that is, the administration of a placebo with full disclosure that it is a placebo. OLP has shown efficacy in chronic non-cancer pain syndromes and has been suggested as a promising candidate for medication tapering. This study aims to assess whether OLPs can enhance the reduction of daily morphine equivalent dose (MED) in chronic non-cancer pain patients and examines its potential in mitigating opioid withdrawal symptoms.
Methods and analysis This study is designed as a randomised, controlled, single-centre trial. Participants will be randomised into either an OLP group or a control group. The study duration will span six to nine weeks, during which all participants will aim to reduce their opioid intake. Both groups will monitor their opioid intake daily using a diary app and will receive feedback on their progress of reducing opioids. Additionally, participants in the OLP group will receive OLP tablets for the entire study period. During the first week, the OLP group will undergo a one week learning phase using a classical conditioning paradigm, where each opioid intake is paired with a placebo. In the subsequent five weeks, the OLP group will enter a dose-extension phase in which only the first opioid intake of the day is paired with a placebo, and additional placebos can be taken as desired. At the end of the study, qualitative interviews will be conducted with the first 15 participants in the OLP group. The primary outcome measure is daily opioid intake. Secondary outcomes include opioid withdrawal symptoms, pain severity, disability, anxiety, depression, opioid beliefs, intervention expectancy and qualitative data. Statistical analyses will include analysis of covariance and regression models.
Ethics and dissemination The ethics committee of the Canton of Zurich, Switzerland, approved the study (SNCTP-nr.: SNCTP000005853/BASEC nr.: 2023–02327).
Participants will be compensated with 100 Swiss Francs for their full participation in the study. Participants who will take part in the qualitative interview will be compensated with additional 15 Swiss Francs.
Trial registration number This study is registered at clinicaltrials.gov: NCT06350786.
- Pain management
- PUBLIC HEALTH
- Chronic Pain
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
This is the first study to assess the effectiveness of an open-label placebo (OLP) intervention for opioid tapering in patients with chronic non-cancer pain using a randomised controlled trial design.
Based on behavioural learning theory, this study uses a conditioning-based learning paradigm to pair opioid intake with placebo intake, potentially inducing the placebo effect.
OLP and the control groups are designed to ensure structural equivalence, with both groups featuring electronic monitoring of opioid intake and the same number of interactions with a member of the study team.
A qualitative assessment is included to explore participants’ experiences with the OLP intervention, providing insights into feasibility and acceptability.
The study is conducted at a single centre, which may limit the generalisability of findings to broader clinical settings.
Introduction
Background and rationale
Chronic non-cancer pain is a major global health issue, affecting nearly 20% of adults in Europe alone.1 This has led to a substantial increase in opioid prescriptions for chronic non-cancer pain management, both in the USA and Europe over the past two decades.2–6 Despite this, there is no consistent international treatment standard for the long-term therapeutic use of opioids in chronic non-cancer pain management, according to the guidelines provided by the Association of Scientific Medical Societies in Germany.7 Additionally, opioid therapy is associated with various adverse events and risks, including abuse, overdose and opioid-induced hyperalgesia.8–10
Since risks often outweigh benefits,11 many patients receiving opioid therapy report the desire to stop or cut down their opioid use.12 13 Current strategies for tapering include dose reduction protocols, pharmacological opioid replacements and non-pharmacological therapies. However, systematic reviews concluded that only low-quality evidence supports the effectiveness of opioid-tapering interventions,9 14 15 while the results show only limited effects.16 The feasibility of non-pharmacological adjunctive therapies is limited in primary care settings as they are typically time-consuming, difficult to access, costly and managed outside the direct patient–physician interaction.17 Pharmacological approaches, on the other hand, lack patient empowerment.18 These challenges prompt the need for developing other types of tapering protocols.
Notably, the endogenous opioid system plays a role in mediating the placebo response, with placebo effects being particularly pronounced in opioid trials.19 20 This is important for chronic non-cancer pain, where patients show substantial and clinically relevant placebo responses.21–23 However, the administration of deceptive placebos violates patients’ right to autonomy.24 25 Administering placebo without deception could harness the placebo effect successfully and ethically.26–29
An open-label placebo (OLP) intervention, that is, the placebo administration with full disclosure of being a placebo, is promising. OLPs have proven effective in managing chronic non-cancer pain syndromes, such as chronic low back pain30 31 and irritable bowel syndrome.22 32 Several (network) meta-analyses have demonstrated that patients receiving OLPs’ experience significantly greater pain relief compared with those in the control groups.33 34 Moreover, and of relevance when it comes to the need to reduce opioid medication, OLPs have been shown to be a promising candidate for medication tapering.35 According to principles of behavioural learning theory, this effect can be understood through a conditioning paradigm, in which the process of medication tapering is facilitated by repeatedly pairing the active medication (unconditioned stimulus) with an OLP (neutral stimulus) during an initial learning phase. Through this repeated association, the body begins to link the intake of the OLP with the effects of the medication. Over time, as medication doses are gradually reduced, the OLP alone takes on the role of a conditioned stimulus, triggering a response similar to that of the medication.36–39 One pilot study with 20 spinal cord injury and polytrauma patients has employed this approach and showed that OLPs are effective.29 In a different study using a single-group design, that examined OLPs as an adjunctive intervention for opioid reduction in acute pain, pain relief scores for placebos and opioids did not differ significantly.28 Furthermore, conditioned OLP has been shown to reduce daily opioid consumption and postsurgical pain among patients recovering from spine surgery: Patients in the conditioned OLP group used approximately 30% less daily morphine milligram equivalents (MMEs) compared with those in the treatment-as-usual group.36 These findings are in line with studies spanning two decades, revealing that it is possible to condition the body’s opioid system.37–40
Despite these promising findings, there is a lack of trials that examine OLP as an adjunctive intervention for the reduction of opioid medication in the chronic non-cancer pain population. This is noteworthy, given that therapeutic opioid doses are associated with side effects, along with clinically relevant responses to OLPs. The OLP intervention offers a promising approach to tapering long-acting opioids within a structured reduction regimen by using a conditioning paradigm that pairs opioid intake with OLP administration. This approach aims to gradually transfer the learned response from the active medication to the OLP.
While prior research has demonstrated the analgesic efficacy of OLPs in chronic pain conditions and their potential in reducing opioid consumption in surgical or acute pain, these findings have not yet been extended to patients with chronic non-cancer pain undergoing structured opioid tapering. Our study addresses this gap by specifically examining the use of OLPs for opioid tapering in the chronic non-cancer pain population. We decided to include a control group attempting opioid reduction without the use of OLPs in order to maintain structural equivalence between groups. Additionally, our study explores the feasibility and acceptability of integrating OLPs into primary care opioid-tapering protocols, areas that remain underexplored in the existing literature.
Aims and objectives
The primary aim of the present study is to evaluate whether the daily morphine equivalent dose (MED) of opioid medication in patients with chronic non-cancer pain can be reduced with an OLP adjunctive intervention compared with a control group. Our secondary aim is to evaluate whether the OLP intervention can reduce opioid withdrawal symptoms in comparison to the control group.
Methods and analysis
Study design
This randomised, controlled, single-centre trial with two parallel groups began in March 2024 and is expected to conclude in early 2026. The study duration will span six to nine weeks, from pre-screening to study conclusion. The primary endpoint is the change in average opioid consumption between the first seven days of the learning phase and the last seven days of the dose-extension phase. It is calculated as mean daily use of opioid medication (ie, the daily MED) over each seven day period. Participants will be assigned to the OLP and control groups. For randomisation, a three-level stratification factor will be determined for the randomisation so that participants will be equally assigned to the OLP and control group, respectively, based on their baseline MED as follows: low (< 40 mg), moderate (40 mg–100 mg), and high (> 100 mg). Participants in both groups will record their daily opioid intake, and the OLP group will additionally record their placebo intake, with the SEMA341 smartphone application. The placebo tablets used in this study (“P-Tabletten blau”, Zentiva) are round, blue, and contain no active pharmacological ingredients. They have been successfully used in previous open-label placebo studies.42 43 The blue colour was chosen based on research indicating that blue is generally perceived as calming and relaxing, which is particularly relevant in the context of pain.44
The data collected in SEMA3 will comprise the name of the opioid, the type of opioid (eg, tablet), daily intake quantities (dosage) as well as time and date of intake. These details will then be used for electronic monitoring feedback, which will be delivered during three visits (seven, 21 and 42 days after baseline). Electronic monitoring feedback will be similar for both groups and will be based on a graph representing daily opioid medication intake, as entered by the participants into SEMA3. During the electronic monitoring feedback, the study member will discuss with the study participant the adherence to their medication intake in an empathic and non-judgmental manner.
Self-reported outcome measures will be collected via surveys on an online platform, Research Electronic Data Capture (REDCap),45 during four visits for both study groups (14 days prior to baseline, at baseline, and seven and 42 days after baseline). Both groups will receive the interventions from the project manager and study team members. All members of the team have been trained in delivering the intervention and have completed the Good Clinical Practice course at the University Hospital Zurich. Due to the transparent nature of the OLP intervention, participants and investigators will not be blinded to group assignments.
Eligibility criteria
Inclusion criteria assessed during the pre-screening procedure include age 18 or older, proficiency in German, chronic non-cancer pain for at least six months, chronic opioid medication use for more than three months, oral intake of opioids, self-reported motivation for opioid reduction, a primary treating physician overseeing the opioid reduction process, and access to a computer or tablet with an email account.
Exclusion criteria include psychotic symptoms, suicidality, cognitive impairment, planned surgery within the next two months, self-reported illicit drug use, harmful alcohol use, intolerance to placebo ingredients (eg, lactose, sucrose, corn-starch), serious health issues preventing study participation, and concurrent involvement in other chronic non-cancer pain studies. Participants may continue to use their regular medication (ie, other than opioid medication).
Recruitment
Participants will be recruited primarily via flyers and posters at the University Hospital of Zurich pain outpatient department and related clinics. Additionally, recruitment will occur at outpatient pain clinics, pain centres, general practitioners' practices, pain community centres, addiction associations, and pharmacies throughout Switzerland, with a focus on the canton of Zürich. Online recruitment will also be conducted. All study processes will take place at the Department of Consultation-Liaison Psychiatry and Psychosomatic Medicine of the University Hospital of Zurich to maintain a single-centre approach.
Study procedures at each visit
For each participant, the study will include a total of six study visits over the course of six to nine weeks. Please refer to figure 1 for a detailed overview of the study design and study flow. Each phase is described below.

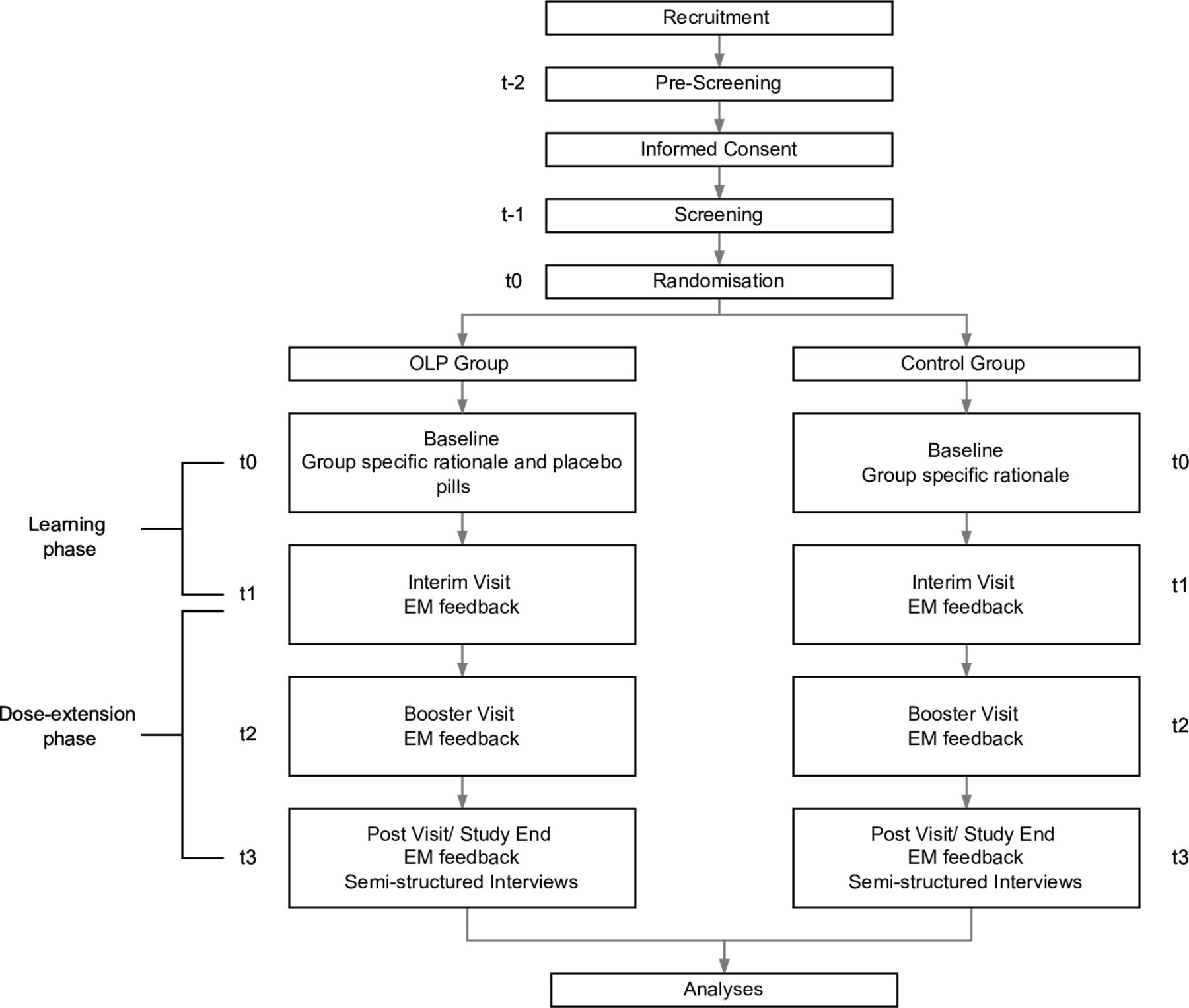{kind=link}
Study Design and Flow of Participants. EM feedback = Electronic Monitoring Feedback.
Pre-screening via telephone call and informed consent (visit 1, timepoint t-2, -21 days prior to baseline)
During an initial telephone contact at timepoint T-2, a pre-screening will take place to verify potential eligibility for participation in the study. Potential study participants will receive detailed study information and the informed consent form by mail.
The original informed consent form can be found in German in the online supplemental material A, as well as an English translation in the online supplemental material B.
Supplemental material
Screening (visit 2, timepoint t-1, -14 days prior to baseline)
Once informed consent is signed by participants, they will complete an online screening questionnaire via REDCap to further evaluate their eligibility. This screening includes more detailed assessments of clinical and demographic criteria compared with the broader criteria evaluated during pre-screening. If eligibility criteria are met, participants will be scheduled for the baseline visit. Additionally, the primary treating physician will be informed about the study participation and will receive a link to a questionnaire in REDCap to assess their expectations regarding whether their patient will reduce the amount of opioids during the study, along with a second questionnaire to evaluate the physician’s acceptability of the OLP approach.
Baseline assessment at study site or via telephone call (visit 3, timepoint t0, 0 days)
Participants will be informed of their group assignment (ie, OLP or control) and will receive a group-specific rationale from a study member. Both treatment rationales will explain the potential benefits of electronic monitoring feedback in supporting patients during voluntary prescription opioid tapering. The treatment rationale for the OLP group is additionally based on the discussion points, adapted from Bernstein,27 including the following statements:
Opioids work by telling your body that you are experiencing less pain.
Placebos should be taken every time an opioid is taken, supporting the reduction of opioid medication (as shown by previous studies).
By pairing the tablets together, your brain will learn to release chemicals like endorphins that cause pain relief in response to the placebo, just as it does in response to the opioid.
At a certain point, placebos might provide adequate pain relief, and you may need fewer opioids.
The OLP group will additionally receive placebo tablets for six weeks (150 placebo tablets=three blister packs).
The treatment rationale for the control group will solely explain the potential benefits of electronic monitoring feedback, without any discussion points, as this group will not receive any placebos.
Subsequently, self-reported baseline surveys will be assessed by both groups online in REDCap. After completing the online surveys, daily monitoring of opioid and placebo tablet (for the OLP group) intake via the SEMA3 app will be explained to the participants.
After the baseline visit, participants in the OLP group will undergo a one week learning phase, where they will pair each opioid intake with an OLP tablet to initiate a learning process via classical conditioning.
Interim visit via telephone call (visit 4, timepoint t1, seven days after baseline)
Seven days after the baseline, a meeting via telephone call will take place between the study participant and a study team member. Participants in both groups will receive a brief repetition of the group-specific rationale. Subsequently, electronic monitoring feedback will be provided to both study groups. Thereafter, the link for the online questionnaires in REDCap will be sent to participants of both groups for the assessment of the secondary outcomes. The same questionnaire as at timepoint t0 will be used.
After the interim visit, the dose-extension phase will start and will continue until the study concludes. In this phase, participants of the OLP group will only pair the first opioid intake of the day with a placebo, regardless of whether it is a long- or short-acting opioid. Pairing the first opioid intake with a placebo helps maintain the conditioned response, in line with the conditioning paradigm. This approach is based on evidence that intermittent reinforcement - continued pairing of placebo with the active drug - enhances resistance to extinction of the conditioned response.46 For the OLP group, additional placebo tablets may be taken throughout the day as needed, and all additional placebo intake will be recorded in the SEMA3 app.
Booster visit via telephone call (visit 5, timepoint t2, 21 days after baseline)
In both groups, a booster meeting via telephone call will take place between the study participant and a study team member to ensure that the intervention has been understood as agreed on in the last meeting, including a brief repetition of the group-specific rationale. Electronic monitoring feedback will be provided to participants in both groups in the same way as during visit 4. For the OLP group, the dose-extension phase will continue unchanged until the end of the study.
Post visit at study site or via telephone call (visit 6, timepoint t3, 42 days after baseline)
At the end of the sixth week, the study intervention will end for both groups, and participants will take part in the last intervention at the study site or via telephone call. Electronic monitoring feedback will be provided to participants in both groups in the same way as during visits 4 and 5. Subsequently, participants will complete an online questionnaire in REDCap with the secondary outcomes and evaluation outcomes.
Furthermore, the first 15 participants of the OLP group will take part in a semi-structured interview. The choice of number was made to gather insights as fast as possible about the chances and hurdles of the OLP approach, also for upcoming trials planned while the current RCT is still ongoing. The qualitative interview will aim to gather in-depth experiences of participants and will cover the following topics (the interview guide can be found in the online supplemental material C):
Experiences of the OLP intervention within the study
Acceptability of the OLP intervention
Prerequisites, ideas, and concerns regarding practical OLP implementation
Patient and public involvement statement
A patient partner was contacted prior to the study start and was involved in detailing the study design and procedures. In a later phase, the patient reviewed the study protocol to ensure clarity and relevance from the patient’s perspective. The patient partner provided valuable input on recruitment strategies, including how to effectively communicate the study to potential participants and make participation more accessible. The patient partner’s involvement also extended to discussions on the structure and feasibility of participation. Additionally, the patient played an active role in simulating the first intervention (t0) and the treatment rationale for the control group, providing feedback regarding the plausibility of the intervention, which helped refine the study approach. The patient partner himself experiences chronic pain and is currently taking opioids, which he aims to reduce. He therefore represents our study population.
Study outcomes
Please refer to online supplemental Table 1 for a detailed overview of study outcomes and measurement time points.
Primary outcome: daily opioid consumption
The primary study outcome will be assessed based on the daily dose of opioid medication consumption. Given that participants will potentially be prescribed a range of opioid medications, daily morphine equivalent dose (MED) will be standardised using the MME conversion factor with the following formula: MED=daily dose of single opioid * MME conversion factor.47 The total morphine equivalents are subsequently summed to assess the total amount of opioid medication taken per day (ie, sum of the different opioids with regard to their daily dose). Each opioid is assigned its own conversion factor in relation to morphine, which can be obtained from conversion tables.47 48
Although the data of the daily intake of the opioid medication will be recorded daily, the two main time points of interest for the primary outcome will be t0 and t3. The change between these two time points will be examined. The average (mean) consumption of opioid medication (MED) during the first seven days (starting with the learning phase) and the last seven days of the dose-extension phase will be calculated for each participant in MED.
Opioid medication capture: all opioid medication intake will be recorded via a survey in REDCap at t0. This will document the exact amount, opioid name, form of administration (eg, tablet, drops), dosage per unit (eg, mg per mL for liquid opioids), frequency of intake and duration of opioid use (ie, how long participants have been taking the medication).
Secondary outcomes
The main secondary outcome is subjective opioid withdrawal symptoms, which will be collected via a survey in REDCap at t0, t1 and t3, respectively. The two main time points of interest are t0 and t3. The change between these two time points will be examined. The Subjective Opiate Withdrawal Scale (SOWS) measures gastrointestinal, autonomic, musculoskeletal and psychiatric symptoms that typically occur during opioid withdrawal.49 In our study, we use the German translation, which was supplemented by four additional questions in the questionnaire.50 The intensity of the withdrawal symptoms is rated by patients on a scale between 0 (not at all) and 4 (extremely)
Further secondary outcomes will be collected via a survey in REDCap. All of these data are collected from the study participants at t0, t1 and t3, respectively, except the intervention expectancy outcome, which will be examined at the study start only (t0). The two main time points of interest are t0 and t3. The change between these two time points will be examined.
Pain severity: pain severity is assessed using the validated German translation of the International Classification of Diseases, 11th Revision (ICD-11) specifiers, also referred to as ‘extension codes’.51 The index combines patient-assessed ratings of pain intensity, pain-related distress and pain-related interference. Each of these dimensions is measured on an 11-point Numeric Rating Scale ranging from 0 (none) to 10 (severe).
Pain disability: the validated German version of the Pain Disability Index (PDI) measures the subjective degree of self-reported impairment caused by the pain problem in everyday life. Seven domains of life are assessed: (1) family and domestic responsibilities, (2) recreation, (3) social activities, (4) occupation, (5) sexual life, (6) self-care and (7) essential activities.52
Anxiety: the validated German version of the Generalised Anxiety Disorder – 7 (GAD-7) is a brief instrument for assessing self-reported generalised anxiety disorder symptoms.53 The seven items of the questionnaire assess the main diagnostic criteria according to the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition diagnoses (DSM-IV) and criteria, including worrying, trouble relaxing and feeling nervous.
Depression: the Patient Health Questionnaire – 9 (PHQ-9) measures the severity of depression in self-report.54 The module consists of nine items and is part of the Patient Health Questionnaire (PHQ-D). The German version was derived from the ‘Prime MD Patient Health Questionnaire’ and is based on the depression criteria of the DSM-IV.54
Beliefs regarding opioid use: the Pain Opioid Analgesics Beliefs – Cancer (POABS-CA) measures pain opioid beliefs based on two components (ie, negative effect beliefs and pain endurance beliefs) with 10 items and a five-point Likert scale ranging from 0 (strongly disagree) to 4 (strongly agree).55 For our study, we use the validated German translation, which has been applied to a primarily non-cancer population.56
Intervention expectancy: at the study start (t0), participants’ intervention expectations will be measured in analogy to our most relevant outcomes. For this, the following item will be used: ‘How confident are you that you can reduce/discontinue your opioid medication?’ Second, to measure the expected withdrawal symptoms at the end of the study, we will use the items from the SOWS.49 The introduction of this questionnaire will be adjusted so that the questions are related to the participants’ expectancy regarding their opioid withdrawal symptoms at the end of the study, rather than their current opioid withdrawal symptoms.
Outcomes related to electronic monitoring
Placebo tablet count: the intake of placebo tablets in the OLP group will be electronically monitored using the SEMA3 app from t0 to t3, serving as the primary way for assessing placebo intake. Additionally, a tablet count of the returned placebo blisters at t3 will be conducted, and the number of remaining tablets will be documented. A ratio can be calculated from the tablet count recorded in the SEMA3 app and the count of tablets from the returned blisters, which can then be used for statistical analysis. For the statistical analysis, the overall placebo intake between t0 and t3 is of interest.
Opioid adherence: Participants in both groups will record their daily intake of opioid medication (including on-demand opioid medication) from t0 to t3 in the SEMA3 app. The data collected this way will be used for electronic monitoring feedback at t1, t2 and t3.
Evaluation outcomes
Rationale credibility: the rationale credibility of the OLP intervention will be assessed in the OLP group at study end (t3). The following questions will be asked: ‘During the study, did you believe that these were placebo tablets that did not contain a pharmacologic agent?’, ‘Did you find the explanation of why the placebo intervention may work helpful?’ and ‘How helpful did you find the explanation of why placebo intervention can work?’. Answers will be rated on a Likert scale ranging from 0 (not at all) to 4 (extremely).
Placebo understanding: the Placebo Understanding Questionnaire will be administered to both study groups at the study end (t3), assessing participants’ understanding of placebo and their attitudes towards non-specific therapies. The first three items of this questionnaire specifically evaluate placebo understanding and will be used for this study.57
Patient–provider connection: the patient–provider connection is a subscale of the validated German version of the Healing Encounters and Attitudes List which can be used independently from the six subscales.58 The seven items are rated on a five-point Likert scale ranging from ‘not at all’ to ‘very strong’ assessing participants’ attitudes towards patient–provider connection as a non-specific intervention effect. The questionnaire will be assessed at study end (t3).
Non-opioid medication intake: participants’ non-opioid medication intake will be assessed via REDCap at t0 and t3 by asking the participants about the medication’s name, dosage and reason for intake.
Intervention implementation: to examine the influence of the individual administering the study intervention, we record at t0, t1, t2 and t3 which study member implemented the intervention in the respective study groups.
Treating physicians’ outcomes
Primary treating physicians’ acceptability of the OLP approach: at t0, the primary treating physicians will be asked about their acceptability of the OLP approach. For this, we ask them to imagine a scenario where a physician uses OLPs to help a patient reduce opioids. We will use two key items: ‘Is it acceptable for the physician to try a placebo intervention?’ and ‘Would you, as a patient, be willing to take this intervention?’ Both questions use a five-point Likert scale ranging from ‘no, not at all’, to ‘yes, totally’.59 Further variables include the perceived competence of the physician, along with patient satisfaction and worry regarding the treatment from the patient’s perspective.12 Competence will be answered on a seven-point Likert scale ranging from ‘incompetent’ to ‘highly competent’. Satisfaction items can be answered on a seven-point Likert scale ranging from ‘very dissatisfied’ to ‘very satisfied’. Worry will be displayed on a seven-point Likert scale ranging from ‘much more worried’ to ‘much less worried’. Finally, four additional items will be asked to assess interpersonal trust in the patient–physician relationship and the doctor’s warmth to be answered on a five-point Likert scale ranging from ‘strongly disagree’ to ‘strongly agree’.60
Primary treating physicians’ intervention expectancies: At t0, the primary treating physicians’ expectations will be measured in analogy to our most relevant outcomes. First, we will ask the physician how satisfied they think that the patient is with the opioid medication and whether they are optimistic that the patient will reduce the amount of opioids. Motivation will be assessed on a satisfaction ruler ranging from 0 % to 100 %.61 Second, to measure their expectation regarding their patients’ withdrawal symptoms at the end of the study, we will use selected items from the SOWS questionnaire.49 The introduction of this questionnaire will be adjusted to ensure that its questions relate to the primary treating physicians’ expectations regarding their patients’ opioid withdrawal symptoms at the conclusion of the study.
Qualitative outcomes
The qualitative interview will be audio-recorded. The study team will create transcripts of the audio recordings using the MAXQDA VERBI software,62 which will be examined subsequently with the method of thematic analysis using the same software.
Additional symptoms
Participants will answer three questions regarding additional symptoms that might have occurred since the last visit. These will be measured during the electronic monitoring feedback at t1, t2 and t3. The questions are the following:
‘How have you been since the last visit?’ (open answer format)
‘Did you have certain symptoms?’ (yes/no)
If the participant answered ‘yes’ to the second question, a follow-up question will be displayed: ‘Please describe the symptoms you had in detail’. (open answer format)
Safety outcomes
A study member will document these additional symptoms and discuss with the project manager or sponsor investigator whether they are related to study participation. If so, they will be recorded as an adverse event in REDCap. Although serious adverse events are not expected due to the nature of the study, potential serious adverse events related to opioid analgesics and their tapering—such as suicidal ideation—may occur. In such cases, the investigator delivering the study intervention will contact the participant’s primary treating physician. If it cannot be excluded that the serious adverse event is attributable to the intervention under investigation, the investigator will report it to the ethics committee within 15 days. Any urgent safety or protective measures taken during the study will also be promptly reported to the ethics committee, as required
Data management
All data and documents will be maintained in one of the following data management systems: (electronic) trial master file, REDCap, and SEMA3. The (electronic) trial master file, monitored by the project manager and study investigator, will ensure restricted access for authorised team members. Person-identifying data of participants will be collected using paper Case Report Forms (pCRFs). Study data for study procedures will be collected and recorded by electronic Case Report Forms (eCRFs) in REDCap. These will be set up by the Clinical Trials Centre of the University Hospital Zurich.
Determination of sample size
A similar study on opioid dose reduction was conducted in chronic non-cancer pain patients, comparing an OLP with a treatment-as-usual group.29 With the data extracted from that study, an SD value of 1.03 could be calculated for the between-group difference.63 This effect size is notably high compared with other OLP studies, with recent meta-analyses reporting overall effects of standardised mean differences 0.7234 and 0.88.64 Given the high variability and exceptional effect size, a conservative effect size of d=0.60 was used for our sample size calculation. The calculation was based on a power of 80% and a significance level (alpha) of 0.05. Using analysis of covariance (ANCOVA) with the allocation group (OLP/control) as an independent between-subject variable, and accounting for a 20% dropout rate, the final sample size is 86 (43 per group).
Statistical analyses
Primary analysis
For the analysis of the primary endpoint, the baseline consumption of opioid medication will be calculated as the average (mean) of the first seven days of the study (from t0). For the analysis, the differences in the consumption of opioid medication across the two groups of this study will be assessed. The primary aim will be analysed by a linear model in an ANCOVA, with the consumption at baseline as covariate and the randomised allocation (OLP vs control group) as explanatory variable. The normality assumption will be graphically assessed. Log transformation will be considered in case of violation. The dependent variable will be the average consumption of the opioid medication over the last seven days at t3. Intention-to-treat analyses will be carried out.
Main secondary analysis
For the analysis of the main secondary endpoint, the differences in the withdrawal symptom scores (SOWS) across the two groups will be assessed. The secondary endpoint will be analysed by a linear model with the withdrawal symptom scores at baseline as covariate and the randomised allocation (OLP vs control group) as explanatory factor (ANCOVA). The normality assumption will be graphically assessed. Log transformation will be considered in case of violation. The dependent variable will be the withdrawal symptom scores at t3.
Other secondary analyses
For the analysis of further secondary endpoints, the differences across the two groups of various other scores assessed during our study will be examined. Each of these endpoints will be analysed by a linear model with their scores at baseline as covariate and the randomised treatment (OLP vs control group) as explanatory factor (ANCOVA). The dependent variable will be the corresponding scores at t3. These secondary endpoints are the following: pain severity degree (ICD-11 specifiers); pain disability degree (PDI); anxiety (GAD-7), depression (PHQ-9); pain opioid beliefs (POABS-CA) and treatment expectancy. Additionally, exploratory sensitivity analyses are planned to examine possible moderators (eg, opioid dosage) of treatment outcomes.65
Analyses for evaluation outcomes
A t-test will be employed to compare the evaluation outcomes between the two groups. In the case that there is no normal distribution for the data, non-parametric procedures such as Wilcoxon/Mann–Whitney will alternatively be used.
Qualitative analyses
We will analyse the qualitative data by means of thematic analysis, taking a hybrid approach of inductive and deductive coding and theme development.66 In this approach, core themes will be first elicited inductively from the collected data.67 Subsequently, the core themes will be examined deductively with respect to theoretical assumptions about the OLP treatment rationale.68
Monitoring
The investigator’s site will collaborate with the Clinical Trials Centre of the University Hospital Zurich to ensure monitoring. Monitoring activities are defined in a study-specific monitoring plan.
Ethics and Dissemination
The study has received approval from the ethics committee of the Canton of Zurich (BASEC-Nr. 2023-02327/SNCTP-Nr.: SNCTP000005853). All participants will provide written informed consent prior to enrolment. Study data will be handled in accordance with national data protection regulations. The results will be submitted for publication in peer-reviewed journals and presented at national and international conferences. Participants may request a summary of the study findings upon completion. The trial is registered at ClinicalTrials.gov (NCT06350786).
Ethics statements
Patient consent for publication
Acknowledgments
We would like to extend our heartfelt gratitude to our students, Tina Maria Daniela Kamer, Massiel Reyes, Susan Singer, Katharina Saurer, Sophie Rosch and Nina Fritschi, who provided assistance during the setup of the study. Their contributions significantly facilitated the execution of this research. Additionally, we would like to thank the Clinical Trials Centre at the University Hospital Zurich for their support and collaboration in the planning and implementation of our study.
References
Footnotes
KC and KB are joint first authors.
KC and KB contributed equally.
Contributors Contributors: KC: conceptualisation, methodology, writing—review and editing. KB: conceptualisation, methodology, writing—original draft preparation, writing—review and editing. AFN: methodology, writing—review and editing. DF: methodology, writing—review and editing. RvK: methodology, writing—review and editing. LB: methodology, writing—review and editing. HK: methodology, writing—review and editing. MB: conceptualisation, writing—review and editing. KS: methodology, writing—review and editing. IA: methodology, writing—review and editing. AJR: patient involvement, writing—review and editing. JR: methodology, writing—review and editing. EO: methodology, writing—review and editing. CL: conceptualisation, methodology, supervision, funding acquisition, writing—review and editing, guarantor of the study.
Funding This work was supported by the Swiss National Science Foundation (SNSF), grant number: PZ00P1_201972
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, conduct, reporting or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.