Article Text

Abstract
Introduction Delirium is a common complication in elderly patients after major surgeries and can lead to poor outcomes such as neurocognitive decline. Acetaminophen is one of the most widely used adjuvants for perioperative multimodal analgesia. Previous studies showed that it can effectively alleviate postoperative pain, promote opioid sparing and exert anti-neuroinflammatory response, showing strong potential for preventing postoperative delirium. We, thus, propose to test the primary hypothesis that postoperative intravenous acetaminophen would reduce delirium over 5 postoperative days in older patients following major non-cardiac surgery.
Methods and analysis We propose a multicentre, randomised, placebo-controlled, parallel-group trial in patients aged>65 years old scheduled for non-cardiac major surgery with general anaesthesia expected to last at least 2 hours. A total of 1930 elderly patients will be enrolled and randomised at 1:1 ratio to acetaminophen or saline placebo groups, stratified by age, education level and trial site with randomsised blocking. Acetaminophen or saline will be given when the surgical suture begins at the end of surgery and, thereafter, a total of seven doses within 48 hours after surgery. Our primary outcome will be the incidence of delirium, assessed two times per day, through the fifth postoperative day. Secondary and exploratory outcomes will include pain scores with movement, total opioid consumption, severity of delirium, intensive care unit and hospital lengths of stay.
Ethics and dissemination This study has been approved by the Ethics Committee of Ren Ji Hospital, Shanghai Jiao Tong University School of Medicine (LY2023-239-C) and approved by each participating centre. This report follows the Consolidated Standards of Reporting Trials reporting guideline for randomised studies. The findings will be shared in academic meetings and peer-reviewed academic journals.
Trial registration number NCT06653465.
- Postoperative Delirium
- Randomized Controlled Trial
- Adult anaesthesia
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
This study includes patients for various types of non-cardiac surgery, the findings of which are more generalisable for clinical practice.
The intermittent boluses of intravenous acetaminophen for 48 hours postoperatively for intervention may lead to positive results.
The long-term follow-up results are mainly based on the patient’s self-assessment questionnaire, which may increase the bias of the results of this study.
Delirium occurring out of visiting hours might be missed.
Introduction
Postoperative delirium (POD)
Delirium is a common complication after major surgeries, manifesting as acute change in mental status, such as inattention, confusion, incoherence and abnormal perceptual function, with a fluctuating course. POD refers to delirium occurring within 7 days after surgery or anaesthesia, primarily within the first 2–3 days.1 POD can lead to a number of adverse outcomes, including a significant increase in mortality, longer hospital and intensive care unit (ICU) stays, accelerated neurocognitive decline,2 reduced quality of life and increased risk of dementia.3
The reported incidence of POD varied from 2.5% to 4.5%,4–7 with a higher prevalence in the elderly. Among patients aged above 60 years old, POD has been reported to occur in 5.0%–50% patients,8–11 varying by surgical procedures. This is mainly because elderly patients often have major chronic comorbid conditions and a decreased physiological reserve to handle the stress of surgery. The variation of POD incidence in studies is attributable to many factors. Most significantly, POD incidence varies among different surgical procedures. For example, delirium is reported in 36%–40% cardiac surgery, neurosurgery and trauma patients,12 while in elective arthroplasty patients, the incidence of delirium is 5%–10%.13 Another major factor in the diversity of reported POD incidence is diagnostic approaches of delirium.14 In cardiac surgery literature, for example, 53% delirium patients were identified with daily mental status assessment and use of a validated diagnostic algorithm,15 while only 9% were reported by interviews with nurses.16 In addition, type of anaesthesia and preoperative cognitive status of the included patients are considered relative to the variation of POD frequency.8
Risk factors and pathophysiology
The pathophysiology of delirium has not been fully understood. Numerous mechanisms are speculated to be related to the occurrence of delirium, among which there is growing evidence showing that neuroinflammation plays an important role in POD.17 After surgical trauma, cellular damage triggers endogenous factors known as damage-associated molecular patterns, which activate immune cells, such as neutrophils and monocytes. Activation of these cells contributes to systemic inflammation, which can impact multiple organs, including the brain. In addition, the complement system and coagulation cascade activation, blood–brain barrier opening and glial activation are also key components that drive cognitive deficits.18
The predisposing factors of POD are multifactorial, such as advanced age, dementia or cognitive impairment,19 20 hearing or vision impairment, alcohol abuse21 and underlying diseases22 (such as stroke, diabetes and atrial fibrillation). Geriatric patients, often with pre-existing neurodegeneration apparently or recessively, are subject to impaired blood–brain barrier, neuronal loss, reduced cerebral blood flow and dopamine and serotonin levels, increasing the brain insults caused by systemic inflammation and resulting in a greater POD incidence.6 10 Furthermore, numerous perioperative factors, including postoperative pain, hypoxemia, dehydration and electrolyte disorders, acid-base imbalance, sleep exfoliation and use of opioids and some sedative drugs,23–25 may also increase this risk.
Acetaminophen
Acetaminophen is one of the most widely used adjuvants for perioperative multimodal analgesia. Peripherally, acetaminophen has similar effects with nonsteriodal anti-inflammatory drugs (NSAIDs) in terms of inhibition of cyclooxygenase COX1, COX2 and COX3.26 Acetaminophen targets cannabinoid CB1 receptors in the brain and spinal cord and activates the cascade of capsaicin–cannabinoid CB1 receptor, thus producing an analgesic effect.27
Recently, an intravenous formulation of acetaminophen was introduced in China. This is particularly suitable for perioperative patients requiring rapid pain relief when the oral route is not available. Compared with the equivalent dose of oral formulation, the bioavailability and predictability of intravenous acetaminophen is higher as it bypasses the first-pass metabolism.28
Rationale for study
Overall, acetaminophen can effectively alleviate postoperative pain, promote opioid sparing and exert anti-neuroinflammatory response, showing strong potential for preventing POD. Indeed, a randomised-controlled trial of 120 older patients undergoing cardiac surgery shows that intravenous acetaminophen reduced delirium by 64%.29 In a retrospective study of 123 geriatric patients over 60 years old with hip fragility fracture, the incidence of delirium was lower with intravenous than oral acetaminophen (15% vs 33%).30 31 However, the relatively small sample size from a single centre and restrictive inclusion limit the generalisability and reliability of acetaminophen application on POD prevention. Furthermore, literature on POD often separates cardiac surgery from non-cardiac surgery32 33 because both incidence and severity of POD are more profound following cardiac surgery with cardiopulmonary bypass, which is presumably related to this distinct surgical procedure.34 Thus, we focus on non-cardiac surgery and propose this multicentre, randomised, parallel-group, controlled clinical trial for maximum applicability to test the hypothesis that postoperative intravenous acetaminophen would reduce POD in older patients following major non-cardiac surgeries.
Methods and analysis
Study overview
This multicentre, randomised, placebo-controlled, parallel-group, controlled trial was designed to study the effect of intravenous acetaminophen on the incidence of POD in older patients (≥65 years) scheduled for major non-cardiac surgeries with general anaesthesia. The trial flow diagram is shown in figure 1. Recruitment is expected to start on 1 June 2025 and finish by 30 June 2027.

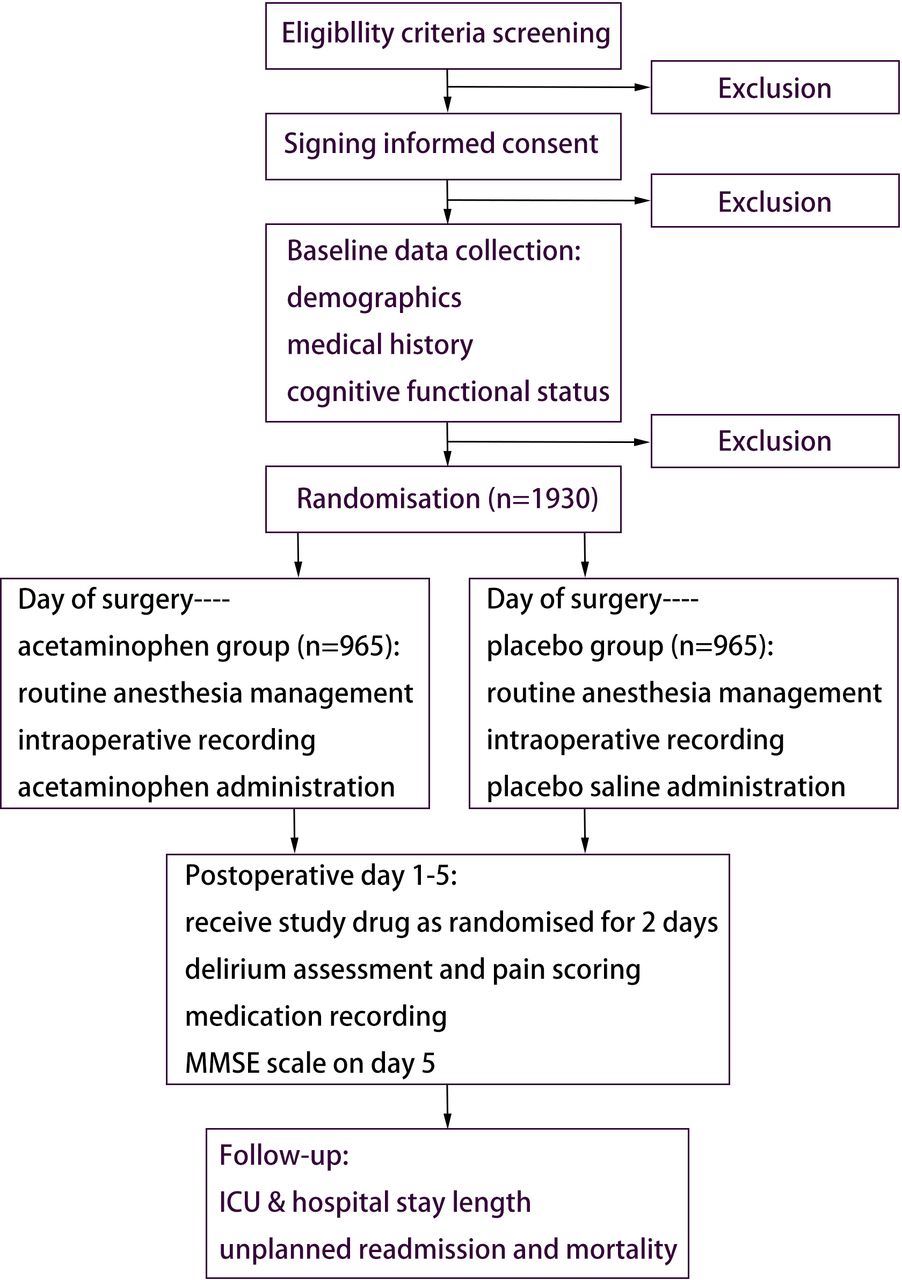{kind=link}
Study flowchart of participant enrolment, intervention, assessment and follow-up. ICU, intensive care unit; MMSE, mini-mental state examination.
Eligibility criteria
Inclusion criteria
Age ≥65 years.
Scheduled for non-cardiac major surgery with general anaesthesia that is expected to last more than 2 hours.
American Society of Anesthesiologists (ASA) physical status I–III.
Weight>50 kg.
Written informed consent.
Exclusion criteria
Pre-existing neuropsychiatric diseases (Alzheimer’s disease, Schizophrenia, Parkinson’s Disease, Seizures, etc).
Pre-existing cognitive impairment (mini-mental state examination (MMSE)<18).
Preoperative delirium.
Severe circulatory instability (preoperative left ventricular ejection fraction less than 30%, unstable angina, severe coronary artery disease, sick sinus syndrome, etc).
Contraindication to the acetaminophen use, including hypersensitivity to acetaminophen,35 severe hepatic impairment (aminotransferase more than three times the upper limit of normal)36 or serum creatinine>177 µmol/L.37 38
Alcohol or drug abuse within a year before surgery.
Inability to communicate because of severe visual/auditory dysfunction or language barrier.
Withdrawal criteria
Patients are always free to withdraw from study at any time. Patients will also be removed from study at any time for adverse events or deemed necessary for patient safety.
Termination criteria
Severe adverse events (SAEs) occur that could not be rescued or cause permanent damage including but not limited to organ failure, significant disability and endangering life and death during the study.
Serious errors are found in the study protocol.
Serious funding or management issues are identified.
The study is cancelled by trial leadership or hospital or regulatory authorities.
Study termination may be short lived or permanent. If the study is terminated early, a written report should be submitted to the Ethics Committee. All case report forms (CRFs) and recorded files should be kept for reference. The resumption of research will require the approval of an ethics committee.
Recruitment
Recruitment advertisement will be posted at the admission office in each participating hospital, on which the purpose, overall design and eligibility criteria of the clinical trial are briefly introduced and contact information is stated for voluntary patients to contact the investigators. Anaesthesiologists trained before surgery and authorised by key researchers will also select potential subjects according to inclusion and exclusion criteria and invite them to participate in this trial during preoperative visits. We plan to recruit patients from 20 participating clinical institutions (online supplemental file 1).
Supplemental material
Informed consent
For every potential participant, investigators must explain the purpose, procedures, potential benefits and risks of the study in easy-to-understand expressions, and give detailed answers to their questions. It must be emphasised to potential participants that he/she has the right to withdraw his/her consent at any time during the study for no reason. Every participant or the authorised surrogate of the participant must sign consent after being given enough time for careful consideration. Online supplemental file 2 shows an example of the participant consent form.
Supplemental material
Randomisation and blinding
Centre-stratified randomisation with a block size of 4–6 will be done using Research Electronic Data Capture (REDCap) software. Patients will be randomised in a 1:1 ratio to receive either acetaminophen or matching placebo. Randomisation will be stratified based on age (65–74, 75–84 and ≥85 years), education level (illiteracy and primary education, secondary education, university or higher) and trial site.
The results of randomisation generated by the REDCap before surgery will be sealed in sequentially numbered opaque envelopes and kept by the pharmacy. The pharmacists who will not participate in the rest of study prepare the trial drugs. Either 4 g acetaminophen (Jiangsu Hengrui Medicine, Jiangsu, China) that was diluted with normal saline to 400 mL or 400 mL normal saline was contained in identical intravenous infusing pumps.
All investigators, healthcare team members and patients were blinded to the treatment group assignment throughout the study period.
Procedure for unblinding if needed
If unexpected SAEs occur, the researcher should report to the ethics committee and expose the individual subjects according to the standard procedure.
The person in charge of the study will log on to the electronic random system to uncover the blindness of individual subjects and timely record the specific time and reason for the emergency unblinding and the name of the person who carried out the unblinding. When unblinding is necessary for safety reasons, researchers should assure that patient care takes priority.
Anaesthesia protocol
Anaesthesia management will follow pre-established clinical guidelines and institutional protocol at the treating hospital. Patients will not be premedicated. Before induction, three-lead electrocardiography, blood pressure and peripheral oxygen saturation monitoring will be placed on the patient. Anaesthesia will be induced with a combination of drugs, including sufentanil/fentanyl, propofol or etomidate and rocuronium/cisatracurium, and maintained with a volatile anaesthesic, propofol and opioids. Rocuronium or cisatracurium will be given per surgical routine. Ketamine, midazolam, phthidine and non-trial acetaminophen are strictly forbidden throughout the study.
Intraoperative management aims to maintain the bispectral index (BIS) between 40 and 60, blood pressure within±20% of baseline and heart rate between 50 and 100. BIS-guided management is implemented to minimise baseline imbalances across study arms, as anaesthetic depth has been reported to be associated with POD incidence among older patients.39 Intraoperative haemodynamic fluctuation should be avoided to reduce perioperative stroke/cardiovascular risk.40 41 Following the principle above, anaesthesia management is left to the discretion of treating anaesthesiologists at each designated site according to their clinical judgement and hospital protocol.
For the intervention, we adopt an intravenous formulation of acetaminophen named ‘Paracetamol and Mannitol Injection’ (Jiangsu Hengrui Medicine, Jiangsu, China). Following the drug label, 50 mL contains 500 mg of acetaminophen or saline placebo will be administered for the first dose when the surgical suture begins at the end of surgery and thereafter repeated every 6 hours for the following seven doses using automatic intravenous pump, and every bolus of 50 mL will be finished within 10 min. Both groups received opioids and other analgesics according to the standard care practices at each participating clinical centre during initial 48 hours postoperatively. However, extra acetaminophen is not allowed.
Postoperatively, patient-controlled analgesia (PCA) pump filled with 2 µg/kg sufentanil, 10 mg metoclopramide and saline to 100 mL will be used. The background rate was set at 2 mL/h, the bolus dose was 2 mL and the lock-out interval was 15 min. The opioids consumption and bolus times will be recorded.
Data collection and management
Demographic and background information
Baseline information, such as demographic data, diagnosis, medical history, preoperative laboratory tests and physical examination, will be collected after obtaining written informed consent. Specifically, baseline functional status will be assessed by MMSE scales, 3-min diagnostic confusion assessment method (3D-CAM) or CAM-ICU, and delirium rating scale 98 revised (DRS-R-98).
Anaesthesia management information
Data will be obtained from electronic medical records, including: (1) duration of surgery and anaesthesia and the doses of anaesthetic drugs given; (2) fluid balance during surgery, including bleeding, type and volume of blood products transfused (document duration of storage before transfusion), type and volume of fluid infusion; (3) mean blood pressure and heart rate during anaesthesia/surgery and (4) length of stay in postanesthesia care unit (PACU) and adverse effects during recovery.
Follow-up visits
Participants will be followed up two times per day during the first 5 postoperative days.11
Incidence of POD: delirium will be assessed by the trained investigators two times per day, from 08:00 to 10:00 and from 17:00 to 19:00, starting from postoperative day 1 using 3D-CAM42 43 or CAM-ICU44 for intubated patients. Immediately before assessing delirium, sedation or agitation was assessed using Richmond Agitation–Sedation Scale (RASS).45 If the patient was too deeply sedated or unarousable (RASS –4 or –5),46 delirium assessment was aborted and the patient was recorded as comatose. Patients with delirium were classified into three motoric subtypes. Hyperactive delirium was defined when RASS was consistently positive (+1 to +4); hypoactive delirium was defined when RASS was consistently neutral or negative (–3 to 0) and mixed delirium was defined when some RASS scores were positive (+1 to +4) and some RASS scores were neutral or negative (–3 to 0).47 48
Delirium severity: for patients with positive assessment of delirium, DRS-R-98 will be used to evaluate the type and severity of delirium following 3D-CAM or CAM-ICU immediately.49 50
Pain scores: pain scores will be assessed using numerical rating scale one times per day, with 0 meaning ‘no pain’ and 10 meaning ‘the worst pain imaginable’.
Cognitive function: on the fifth day after operation, cognitive function will be re-evaluated with MMSE scale.
Medications: all medications, including sedatives, anticholinergics and analgesics, used during postoperative days 1–5 will be recorded. For opioid use, intravenous fentanyl will be recommended.
Length of stay in ICU and hospital after surgery and all-cause in-hospital mortality will be collected from electronic medical records.
Planned interventions and data collection are presented in table 1.
Study schedule
Data management
Data will be collected electronically by nurses or clinicians blinded to group allocation through mobile terminals, such as mobile phones, tablets or computers. Investigators should promptly, completely and accurately record data in the CRFs according to original observation. Supervisor(s)/study coordinator(s) will monitor the conduct of the study. This process can be realised by REDCap, and the data can be exported for statistical analysis directly. Trial data will be entered at each site into a custom secure REDCap database that will be maintained at the coordinating centre in Renji Hospital. The system will record who accessed the randomisation system, when it was accessed and the designated treatment. The database will include appropriate range checks and track all changes. Raw data will flow from trial sites to Renji hospital, but not among sites. Deidentified individual patient data will be shared as necessary among trial sites to support subanalyses.
Each site will have separate unblinded and blinded investigator teams. Each investigator will personally log into Redcap and only have access to appropriate information. For example, unblinded investigators will know the randomised treatment assignment and enter intervention data. In contrast, blinded investigators will be unaware of the randomisation and will obtain all postoperative data, including ward rescue treatment, delirium, pain score, opioid consumption and all postdischarge information. The eCRFs will include appropriate range checks and track all changes.
Study outcomes
Primary outcome
The incidence of POD during the initial 5 days postoperatively.
Secondary outcomes
Delirium type, severity and duration.
Postoperative pain score and total opioid consumption.
Exploratory outcomes
Length of stay in ICU and hospital after surgery.
Cognitive function on the fifth day after operation.
Statistical analysis
Our primary analysis will be intent-to-treat, with a per-protocol analysis for sensitivity. Two-tailed tests will be used in all statistical analyses, and P values of less than 0.05 will be considered to indicate statistical significance.
Baseline data will be presented according to the treatment allocation in an appropriate way; categorical variables and continuous variables are indicated by quantity (percentage) and means±SD or medians (IQR). We will assess the balance between the randomised groups on baseline variables using absolute standardised differences, the difference in percentages and means and medians divided by the pooled SD. When absolute standardised differences exceed 0.10, those variables will be adjusted in the primary analyses.
The primary outcome of the proposed study is POD, defined as the occurrence of delirium on any POD during admission, as assessed using the CAM or CAM-ICU daily until hospital discharge, usually within 5 days postoperatively. This variable will be defined as either present or absent for our primary analysis. The comparison of our primary outcome will be performed using logistic regression analysis and adjusted as necessary for baseline characteristics. To avoid the impact of opioid use on POD incidence, we also plan to analyse the quantity of opioid use as a mediating factor.
Additional postoperative analgesic requirements will be measured as the amount of morphine or its equivalent dosage of other analgesic required during the initial 5 days postoperatively. This value will be compared between treatment groups. Differences in the overall hospital and ICU length of stay, as well as morphine equivalents, will be compared using parametric or non-parametric t-tests.
Sample size calculation
The sample size was calculated from the assumption that the incidence of POD is about 15% 8 9 among elderly patients not younger than 65 years and a 30% treatment effect (10.5%) of the acetaminophen group was expected to develop POD. 917 participants in each group are required to have 90% power at two-sided 0.05 significance level without interim analyses. With an anticipated 5% dropout rate,51 we concluded that a total of 965 participants for each group are required, 1930 participants totally.
Oversight and monitoring
Coordinating centre and trial steering committee (TSC)
Renji Hospital, Shanghai Jiao Tong University School of Medicine is the coordinating centre, who will convene TSC, including as a minimum an independent chair, independent clinician, the chief investigator and trial manager. The role of the TSC is to provide the overall supervision of trial conduct and progress. Details of membership, responsibilities and frequency of meetings will be defined in a separate charter.
Data monitoring committee (DMC)
The DMC, composed of principal investigators from each centre, statisticians and representatives from the ethics committee, will be responsible for data monitoring. Members of the DMC are independent of the sponsors and other stakeholders. The role of the DMC will include the following: monitoring the data and making recommendations to the TSC on whether there are any ethical or safety reasons why the trial should not continue, considering the need for any interim analysis, advising the TSC regarding the release of data and/or information and considering data emerging from other related studies. Written reports on trial progress and feedback will be submitted to the committee quarterly, and the cases of unexpected scenarios and SAEs will be discussed at committee meetings.
Adverse events managements
Possible adverse events include anaesthesia-related adverse events and perioperative analgesia-related adverse events. Acetaminophen-related adverse events include acute liver failure36 (total bilirubin >10 time the upper limit of normal or daily increase 17.1 µmol/L; prothrombin activity <40% or international normalised ratio >1.5 and hepatic encephalopathy and exclude other causes within 2 weeks) and allergic reaction,35 which could be characterised by skin itching, rash, dyspnoea and laryngeal oedema. The occurrence of adverse events will be monitored from the beginning of anaesthesia until 5 days after surgery.
Management and documentation of adverse effects
We will limit the scope of our adverse event monitoring and reporting to all serious adverse events, including unexpected death, believed to be related to the study intervention as well as non-serious adverse events believed to be possibly or probably. Any adverse event (AE) should be documented in the corresponding section of the case report form, including time of occurrence, diagnosis, time of diagnosis, management, duration, sequelae and severity. Any adverse events should be treated promptly according to routine practice and should be followed up until they have completely resolved. The date of cessation of AE or AE-related symptoms must be tracked. If the AE still exists, do not fill in the end date. SAEs are defined as events that cause serious harmful consequences in the course of clinical trials, such as prolonged hospitalisation, disability, affecting working ability, endangering life or death and causing organ failure.
When SAEs occur, we should make every effort to rescue and actively take various measures to avoid permanent damage. The SAE investigator must notify the applicant, clinical trial organisation and ethics committee by telephone or fax within 24 hours from the beginning of the trial to the observation period and fill in the section of the case report form on serious adverse events. The SAE’s medical information should also be reported to the applicant after 24 hours.
All SAEs need to be reported within 48 hours of knowledge of the event to the Project Office. For such events, research personnel will complete an SAE CRF in the database. The Project Office will then inform regulatory authorities in a timely manner, as necessary, according to the applicable regulations.
The DMC will provide oversight of patients’ safety throughout the trial by reviewing aggregate data (including all reported study outcome events and SAEs) by treatment group at regular intervals throughout the duration of the trial and as defined in the DMC Charter.
Patient and public involvement
Patients and/or public were not involved in the design of this protocol.
Discussion
Significance
With the development of society and the progress of medical technology, the increase in the number of geriatric surgeries is attracting worldwide attention. For elderly people, POD is a prevalent and serious threat. Reducing the risk of POD has always been a complex issue, and the investigators have done a lot of research on this subject. Previous studies have investigated perioperative interventions, including the use of dexmedetomidine,29 32 combined nerve tissue and inhalation anaesthesia,5 52 to reduce the incidence of POD, but the effect is still unclear. Acetaminophen is likely to prevent POD by effectively reducing the use of opioids.31 As far as we know, this is the first multicentre randomised-controlled clinical trial to investigate intravenous acetaminophen as prevention for POD in elderly patients scheduled for non-cardiac surgery. This study will test whether acetaminophen could reduce the incidence of POD in elderly patients in terms of providing insight into postoperative pain and long-term cognitive functional status. We expect that the findings of the study will finally add to the current understanding of POD in major surgery and contribute to the international recommendations of geriatric surgery management.
Limitations
This study has several noteworthy limitations. First of all, the long-term follow-up results are mainly based on the patient’s self-assessment questionnaire, which may increase the bias of the results of this study. Second, we cannot treat all patients in a unified way. Factors, such as anaesthesia, use of analgesics, prophylactic use of antibiotics and steroids, intraoperative environmental management and postoperative treatment measures, will be determined according to the best judgement of clinicians. Randomisation stratified by trial site would be expected to limit clinical practice differences between study groups. Third, given the fluctuating nature of delirium, some patients with delirium onset at other times might be missed. Despite this, assessment two times a day for 5 postoperative days has been adopted for POD detection in previous research.39 Fourth, males have been suggested to have a higher risk of POD in recent studies.53 54 Gender bias will be adjusted for baseline balance if necessary when comparing POD incidence between the two factorial groups. Other factors that might impact POD, including the type and duration of surgery, will also be evaluated in statistical analysis.
Ethics and dissemination
This study has been approved by the Ethics Committee of Ren Ji Hospital, Shanghai Jiao Tong University School of Medicine (LY2023-239-C), and approved by each participating centre. The trial was registered at ClinicalTrials.gov with the registration identifier (NCT06653465) on 20 October 2024. This report follows the Consolidated Standards of Reporting Trials reporting guideline for randomised studies. The findings will be shared in academic meetings and peer-reviewed academic journals.
Trial status
This study was approved by the Ethics Committee of Ren Ji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China, on 26 December 2023 (LY2023-239-C). It was registered at ClinicalTrials.gov on 20 October 2024 (NCT06653465). Recruitment is expected to start on 1 June 2025 and is expected to finish by 30 June 2027.
Ethics statements
Patient consent for publication
Acknowledgments
We acknowledge the effort and support by all personnel involved in this trial.
References
Footnotes
ZW and LY are joint senior authors.
MZ and BW are joint first authors.
MZ and BW contributed equally.
Contributors LY and ZW conceived and designed the study. MZ and BW drafted and revised the manuscript of this protocol and contributed equally to this article. MM and YW criticised and revised the manuscript. LY and ZW overviewed the manuscript. All authors reviewed and approved the publication of the final version of this protocol. The guarantor of this study is LY, who accepts full responsibility for the design, conduct and publication of the study.
Funding This study is supported by the National Natural Science Foundation of China (No. 82371517, U23A20508), Pudong New Area Health Commission Research Project (No. PW2022D-01), the Fundamental Research Funds for the Central Universities (No. 24X010202059), and Shanghai Engineering Research Centre of Peri-Operative Organ Support and Function Preservation (No. 20DZ2254200).
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.