Article Text

Abstract
Introduction Residual thrombosis risk is an important contributor to ischaemic events in patients with acute coronary syndrome (ACS) after percutaneous coronary intervention (PCI). Although previous studies have shown that rivaroxaban 2.5 mg two times per day in ACS patients with high ischaemic risk can significantly reduce the risk of ischaemic recurrence and mortality, individualised treatment with low-dose rivaroxaban is still rare. Using D-dimer and PARIS (Patterns of non-Adherence to Anti-Platelet Regimen in Stented Patients) coronary thrombosis risk score to identify ACS patients at high ischaemic risk, we aim to investigate whether 3-month low-dose rivaroxaban therapy on the basis of dual antiplatelet therapy (DAPT) could result in reduced ischaemic events without increasing bleeding.
Methods and analysis This study is a multicentre, prospective, open-label, randomised controlled trial involving 3944 ACS patients undergoing PCI from more than 40 tertiary hospitals in China (ClinicalTrials.gov NCT05638867). Patients with PARIS coronary thrombosis score ≥3 or D-dimer ≥0.28 µg/mL will be 1:1 randomised to the experimental group (rivaroxaban 2.5 mg two times per day for 3 months on the basis of 1 year standard DAPT) or the control group (1 year standard DAPT only). The primary endpoint of this study is major adverse cardiovascular and cerebrovascular events (MACCE), a composite of death, myocardial infarction, unplanned ischaemia-driven revascularisation and systemic embolic events. The safety endpoint is Bleeding Academic Research Consortium (BARC) type 3 and 5 bleeding events.
Ethics and dissemination Institutional Review Board (IRB) approval by the Ethics Committee of Fuwai Hospital was obtained on 22 April 2023 (Approval No. 2023–1980). The investigators will have access to the final dataset. Trial results will be made public through publication in professional journals.
Trial registration number NCT05638867.
- Coronary heart disease
- Coronary intervention
- Research Design
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Strengths and limitations of this study
Open-label, multicentre, randomised controlled trial.
Evaluate the efficacy and safety of short-term triple antithrombotic therapy in comparison with standard dual antiplatelet therapy, in ACS patients with high thrombotic risk.
Long-term follow-up for up to 1 year.
Multiple clinical endpoints including net adverse clinical events.
With the open-label design, patients are aware of the medication they are assigned to, which may introduce a bias due to the possibility of not taking rivaroxaban as required. Patients receiving ticagrelor will not be included.
Background
Despite successful revascularisation and effective secondary prevention, the rate of recurrent ischaemic events remains high for some patients after percutaneous coronary intervention (PCI).1 Residual thrombotic risk is an important contributor to ischaemic events in patients with acute coronary syndrome (ACS), in which thrombin plays an essential role.2 The clinical challenge of balancing ischaemic and bleeding risks complicates efforts to optimise anticoagulation therapy for ischaemic event prevention. Clinical evidence suggests that, despite an elevated bleeding risk, the novel oral anticoagulant (NOAC) rivaroxaban demonstrates efficacy in reducing ischaemic recurrence, cardiac mortality and all-cause mortality in patients with coronary artery disease (CAD), thereby yielding a net clinical benefit compared with conventional therapies.2–4
Identifying high-risk groups through coronary thrombosis scores and biomarkers enables individualised antithrombotic regimens with NOACs, thereby reducing future ischaemic events. D-dimer is a highly sensitive biomarker for thrombosis, elevated levels of which reflect enhanced coagulation and fibrinolytic activity.5 6 Our prior study demonstrated that a baseline D-dimer level ≥0.28 µg/mL was independently associated with both 2 year all-cause mortality and cardiac mortality.7 Using the Patterns of non-Adherence to Anti-Platelet Regimen in Stented Patients (PARIS) coronary thrombosis risk score as the basic model, we found that including D-dimer ≥0.28 µg/mL in the score can significantly improve the predictive power over 2 year cardiac death and coronary thrombosis events (CTEs).8
By combining the PARIS coronary thrombosis risk score and D-dimer, we can better identify suitable candidates for short-term triple antithrombotic therapy (TAT) with rivaroxaban and dual antiplatelet therapy (DAPT). With the aim to reduce ischaemic events without increasing severe bleeding, we conducted this multicentre randomised controlled trial (RCT) to evaluate the efficacy and safety of short-term TAT in patients with ACS at high thrombotic risk.
Methods
Trial design
The PARIS Coronary Thrombosis Risk Score Combined With D-dimer to Guide New Oral Anticoagulant Antithrombotic Therapy in Patients With Acute Coronary Syndrome After Percutaneous Coronary Intervention (PRIDE-ACS) trial is an open-label, multicentre, randomised controlled trial. The objective is to evaluate the efficacy and safety of short-term TAT (low-dose rivaroxaban plus DAPT) in comparison with standard DAPT, in ACS patients with PARIS coronary thrombosis score ≥3 or baseline D-dimer ≥0.28 µg/mL.
Study setting and eligibility criteria
A total of 40 hospitals from China are expected to participate in the current study. Key inclusion criteria include (1) patients presenting with ACS, (2) underwent successful PCI and (3) PARIS coronary thrombosis risk score ≥3 points or a baseline D-dimer ≥0.28 µg/mL. Key exclusion criteria include (1) platelet <90×106; (2) haemoglobin <110 g/L; (3) creatinine clearance <30 mL/min at the time of screening; (4) clinically significant gastrointestinal bleeding 12 months before randomisation; (5) previous intracranial haemorrhage, aneurysm, cerebral vascular malformation, etc. (6) history of ischaemic, haemorrhagic stroke or transient ischaemic attack (TIA); (7) severe hepatic insufficiency and (8) indications for anticoagulation, that is, atrial fibrillation, mechanical valve, deep vein thrombosis. Detailed inclusion and exclusion criteria are listed in supplementary files (online supplemental table 1). Patients are enrolled after a preliminary screening to confirm all inclusion and exclusion criteria are obeyed.
Supplemental material
Allocation and randomisation
All subjects will be screened according to unified inclusion/exclusion criteria. All participants will be required to sign an informed consent form (see online supplemental file 2) before enrolment. Randomisation of patients to the experiment or the control group at a 1:1 ratio will be performed with the use of an Interactive Web Response System (IWRS), which will independently generate and handle the randomisation schedule, and treatment allocation will not be known by any investigators in advance. Through the IWRS randomisation process, all major risk factors for the endpoints of this study will be automatically balanced between the two groups.
Supplemental material
Intervention and control
All patients receive long-term or loading doses of DAPT before PCI. The regimen is aspirin 75–100 mg once per day for at least 3 days and clopidogrel 75 mg once per day for at least 6 days before PCI. Patients who are not on long-term DAPT will still be eligible after receiving a loading dose, which contains aspirin 300 mg and clopidogrel 300 mg before PCI, followed by aspirin 75–100 mg and clopidogrel 75 mg orally once per day. For patients presenting as ST elevation myocardial infarction indicated for urgent PCI but not previously on DAPT, they will be screened for eligibility after loading dose DAPT administration and stabilisation of initial symptoms.
Patients assigned to the experiment group will receive TAT for the first 3 months (aspirin 75–100 mg once per day, clopidogrel 75 mg once per day and rivaroxaban 2.5 mg two times per day), followed by conventional DAPT for 9 months. Patients assigned to the control group will receive conventional DAPT for 1 year. During follow-up, the basic DAPT regimen of the two groups remains unchanged. D-dimer level will be re-examined at 3, 6 and 12 months (figure 1).

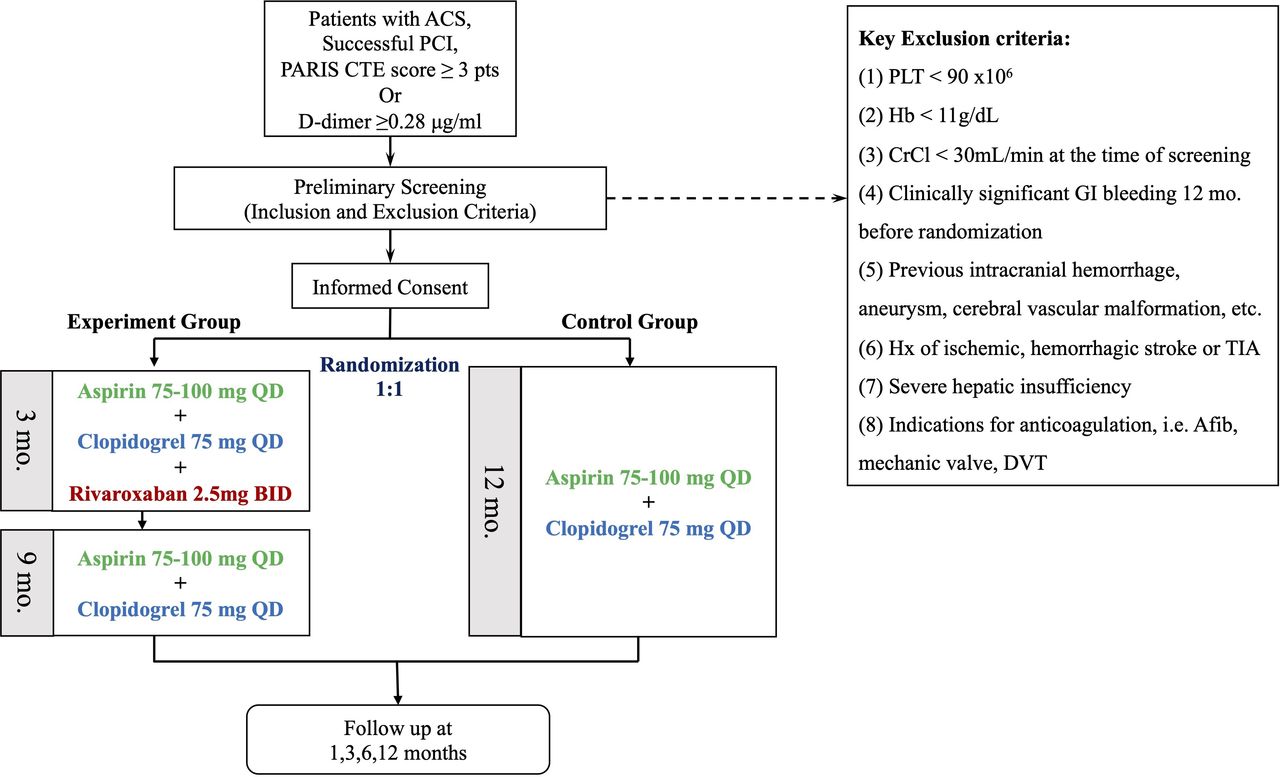{kind=link}
Patient flowchart. ACS, acute coronary syndrome; Afib, atrial fibrillation; BID, twice per day; CrCl, creatinine clearance; CTE, coronary thrombosis events; DVT, deep vein thrombosis; GI, gastrointestinal; Hb, haemoglobin; PARIS, Patterns of non-Adherence to Anti-Platelet Regimen in Stented Patients; PCI, percutaneous coronary intervention; PLT, platelet; QD, once per day; TIA, transient ischaemic attack.
Participant timeline
Baseline physical examination results, medical history, ECGs, laboratory testing results, as well as concomitant medications will be collected on admission according to supplementary files (online supplemental table 2). Clinic follow-up visits are scheduled at 3 months (±1 week), 6 months (±1 month) and 12 months (±1 month). Coagulation function test (including D-dimer), complete blood count, urinalysis, stool analysis, biochemical test and biomarkers test will be re-checked at 3 month and 6 month follow-up. Patients with significantly abnormal test results (abnormal liver/renal function, haemoglobin decrease, etc) during follow-up will be reviewed by the physician for a safety check and, if necessary, be withdrawn from the study. These interim test results may also be used for secondary analysis of the outcomes. Evaluation of compliance, concomitant medication and documentation of adverse events will be performed during each follow-up. Enrolled patients were told to contact investigators should they experience adverse events, and investigators will provide necessary help to aid in their treatment. Regular telephone follow-up will also record endpoints that happened at home and other hospitals.
Outcomes
The follow-up period of this study is 12 months. The primary endpoint is major adverse cardiovascular and cerebrovascular events (MACCE), a composite of all-cause death, myocardial infarction, stroke, unplanned ischaemia-driven revascularisation and systemic embolism composite events. Secondary endpoints included all-cause death, cardiac death, myocardial infarction, stroke, unplanned ischaemia-driven revascularisation, stent thrombosis and systemic embolism. The safety endpoint is Bleeding Academic Research Consortium (BARC) 3, 5 bleeding events. Detailed definitions of the endpoints are listed in the supplementary files (online supplemental table 3).
Power calculation
The power calculation of this study is performed using SAS 9.4 software. With reference to previous studies, we expect the incidence of MACCE events at 12 months to be 13% and 10% in the control group2 9 and experiment group,2 9 10 respectively. The recruitment of patients is expected to last for 3 years, and follow-up will continue for 12 months after the recruitment is completed. Based on this assumption and the superiority study design, an estimated sample size of 1774 in each group will give 80% (two-sided α=0.05 and β=0.20) statistical power to detect a significant difference. Considering a dropout rate of 10%, the total target sample size is set at 3944 patients (1972 in each arm) to meet the statistical power required.
Data management
Designated clinical research coordinators will be responsible for data entry, coding, security and storage through an electronic data capture system. Data quality is ensured through range checks for data values by the investigators. Personal information about potential and informed participants will be anonymised and protected at all times.
Statistical methods
For patient baseline data, categorical variables will be presented as frequencies and percentages. Missing baseline values will be imputed using the multiple imputation method, where applicable. Continuous variables will be displayed as mean±SD or median (IQR), as appropriate. Continuous variables will be analysed for normality using the Kolmogorov-Smirnov test. χ2 test or Fisher’s exact test will be used in comparisons of categorical variables between groups, whereas the Student’s t-test or Wilcoxon rank-sum test will be used in the comparison of continuous variables, as appropriate. First-time event rates will be estimated for each group using the Kaplan-Meier method and compared by the log-rank test. Patients who are lost to follow-up will be censored at the last available contact. Differences between groups will be estimated by HRs with 95% CIs using Cox proportional hazards models. The time-dependent Cox regression model will be applied when the Cox proportional hazards assumption is violated. Baseline covariates to be adjusted for in the Cox regression models include age, gender, other risk factors (Body Mass Index (BMI), smoking), comorbidities (hypertension, diabetes, prior PCI, prior coronary artery bypass graft surgery, heart failure) and key results (including left ventricular ejection fraction, the severity of CAD reflected by SYNTAX score), etc. A two-tailed p <0.05 will be considered statistically significant. Statistical analysis will be performed using SPSS V. 24 software (or higher version; SPSS).
The primary analysis will be performed according to the intention-to-treat principle. Primary and major secondary endpoints will also be analysed in per-protocol and as-treated populations. The per-protocol population consists of patients who were successfully randomised and underwent the assigned treatment, excluding those with major protocol deviations. The as-treated population consists of patients who are analysed according to the actual strategy used for treatment rather than their randomisation assignment.
Additionally, the PRECISE-DAPT score11 will be calculated for all patients retrospectively for quantitative bleeding risk assessment. Secondary analysis of primary, secondary and safety endpoints will be performed based on PRECISE-DAPT score. Using D-dimer as a thrombotic marker,12 repeated results at 3 and 6 months will be analysed to study whether the fluctuation of D-dimer is associated with ischaemic events as a secondary analysis.
Ethics approval and protocol amendments
Institutional Review Board (IRB) approval by the Ethics Committee of Fuwai Hospital was obtained on 22 April 2023 (Approval No. 2023–1980). The approved study protocol, informed consent and other relevant materials are sent to other participating centres. Before patient enrolment and randomisation, all centres should gain approval from their own ethics committee. The study protocol was registered on ClinicalTrials.gov (no. NCT05638867) in November 2022. If important protocol modifications are necessary, all investigators, IRBs of all participating centres and relevant regulators will be notified.
Patient and public involvement
Patients and/or the public were not involved in this study.
Issues concerning the Steering Committee, Clinical Events Committee, Data Safety Monitoring Board, concomitant medications, Adverse Events and Suspected and Unexpected Serious Adverse Reactions are listed in detail in the online supplemental file.
Discussion
Even after successful revascularisation and effective secondary prevention, recurrent ischaemic events still occur in 5–10% of patients after PCI annually.1 In long-term follow-up cohort studies, we found that rates of recurrent ischaemic events are even higher in patients with triple-vessel CAD13 or non-ST elevation ACS (NSTE-ACS).14 Since residual thrombosis risk stands out among various causes of ischaemic events after PCI, reducing residual thrombosis risk is crucial in improving patient prognosis.
In addition to platelets, thrombin also plays an essential role in the process of thrombus formation, the activity of which continues to rise for more than 6 months after ACS.2 In CAD patients indicated for anticoagulation such as atrial fibrillation, several randomised controlled trials have investigated the efficacy and safety between different triple antiplatelet therapies and alternative antithrombotic regimens (dual pathway inhibition or low-dose rivaroxaban plus DAPT).10 15–18 But in ACS patients without other anticoagulation indications, proper anticoagulation therapy to reduce ischaemic events has been under heated debate.
Rivaroxaban, a selective factor Xa inhibitor, inhibits thrombin formation by blocking both intrinsic and extrinsic coagulation cascades. The Anti‐Xa Therapy to Lower cardiovascular events in addition to Aspirin with or without thienopyridine therapy in Subjects with Acute Coronary Syndrome – Thrombolysis in Myocardial Infarction (ATLAS ACS 2-TIMI 51) study4 found that in patients with ACS, rivaroxaban 2.5 mg two times per day brings a clear net clinical benefit by significantly reducing the risk of ischaemic recurrence, cardiac death and all-cause death. Recent European guidelines on STEMI and NSTE-ACS both recommended that low-dose rivaroxaban (2.5 mg two times per day) can be considered in combination with aspirin and clopidogrel for patients at low bleeding risk (IIb B). However, using low-dose rivaroxaban in ACS patients is still debatable, and there still lacks evidence on the optimal duration of TAT as well as how to identify high-ischaemia-risk patients.
The ATLAS ACS2-TIMI51 study confirmed that TAT (DAPT combined with rivaroxaban) significantly reduces ischaemic risk, but at the cost of increased non-coronary artery bypass graft (CABG)-related TIMI major bleeding.2 On the primary efficacy endpoint (ischaemic events), the cumulative incidence curves of the rivaroxaban and placebo groups began to separate at 30 days during follow-up, after which the curves seemed parallel. In terms of safety endpoints, compared with TAT, the risk of major bleeding at 3 months was significantly lower than that at 6 months in the DAPT group (0.5% vs 0.8%).2 In other words, longer-term TAT beyond 3 months seems ineffective in decreasing thrombotic events but contrarily might bring additional bleeding risk. Therefore, in a population with high ischaemic risk, shorter-duration TAT might be promising in improving net clinical outcomes. Based on the results from the ATLAS ACS2-TIMI51 trial,2 the ATLAS ACS-TIMI46 trial3 and analysis from our own cohort of Chinese PCI patients,8 we have limited TAT candidates to high-thrombotic-risk ACS patients and shortened TAT duration to 3 months in designing this trial.
It is worth noticing that in this trial, the P2Y12 inhibitor used in ACS patients is clopidogrel, rather than guideline-recommended third-generation P2Y12 inhibitors, ticagrelor or prasugrel. Clopidogrel is chosen over ticagrelor or prasugrel for the following considerations: First, in the East Asian population, ticagrelor or prasugrel showed no ischaemic benefits but increased bleeding risk compared with clopidogrel in the KAMIR-NIH study19 and PHILO trial.20 A recent meta-analysis of eight RCTs found that in the East Asian populations, ticagrelor is associated with a significantly increased incidence of any bleeding, PLATO major bleeding and dyspnoea in ACS patients compared with clopidogrel.21 Second, to avoid increased bleeding risk, the P2Y12 inhibitor was clopidogrel in most previous trials on TAT. Due to limited evidence, the 2023 European Society of Cardiology (ESC) guidelines on ACS did not recommend ticagrelor or prasugrel use in ACS patients indicated for oral anticoagulation.22 Third, prasugrel is not available for routine clinical use in China.
D-dimer is a specific degradation product of cross-linked fibrin and a highly sensitive biomarker of thrombus formation.5 In recent years, multiple studies have shown that D-dimer levels are associated with long-term adverse cardiovascular events in patients with ACS and stable CAD.12 23 24 Nevertheless, due to low specificity, D-dimer alone has limited power in risk stratification and prognosis assessment in ACS patients.
Compared with a single biomarker, scoring models can better identify high-risk patients and predict short-term and long-term adverse events. Currently, the PARIS coronary thrombosis risk score recommended by the guidelines is a common scoring tool used to predict the risk of ischaemic adverse events in patients taking DAPT after PCI.25 However, validation studies, including the original validation cohort25 and our own cohort,7 26 revealed modest predictive ability for thrombosis-related endpoints. One of the limitations of the PARIS coronary thrombosis score is that no biomarkers are incorporated into the scoring system to predict thrombotic events.
Preliminary results of our cohort study showed that in patients with PARIS coronary thrombosis score ≥3 points, adding D-dimer ≥0.28 μg/mL can improve the predictive value of PARIS coronary thrombosis score for 2 year cardiac death and CTEs (C-statistics increased to 0.721 and 0.655, both p <0.05), with a significant improvement in net reclassification index (p <0.05).8 Furthermore, in patients with PARIS coronary thrombosis score ≥3 points, patients with D-dimer ≥0.28 μg/mL had a low and similar rate of BARC type 3 and 5 bleeding compared with the D-dimer <0.28 μg/mL counterparts (0.6% vs 0.5%, p >0.05).8 It seems combining D-dimer and PARIS coronary thrombosis score can better identify ACS patients at high thrombosis risk, the ideal population for precise short-term TAT with rivaroxaban.
In conclusion, by combining the PARIS coronary thrombosis risk score and D-dimer, the current study aims to identify ACS patients at high thrombosis risk after PCI and investigate whether 3 month TAT with low-dose rivaroxaban could reduce ischaemia without increasing severe bleeding. The results of this study are expected to provide strong evidence for rivaroxaban therapy in selected high-risk ACS patients after PCI.
Limitation
First, due to the open-label design, investigators were not blinded to treatment allocations; therefore, potential bias during outcome assessment cannot be excluded. Second, randomisation across all participating centres was handled centrally without a centre-stratified approach. However, as all participating centres are large tertiary hospitals with similar standards of medical care, the likelihood of centre-based bias is reduced. Third, patients receiving ticagrelor were excluded from the study.
Trial status
Recruiting.
Current study protocol: V.2.0. 27 December 2023.
Recruitment began on 25 November 2023.
Expected date of completion: 31 December 2026.
Protocol amendment since trial commencement: Due to slow recruitment, the investigators proposed an amendment to the third inclusion criteria from ‘Elevated D-dimer levels (≥0.28 µg/mL) on admission AND PARIS coronary thrombosis risk score ≥3 points’ to ‘Elevated D-dimer levels (≥0.28 µg/mL) on admission OR PARIS coronary thrombosis risk score ≥3 points’, on 27 December 2023. The IRB reviewed and approved the protocol amendment on 12 January 2024.
Ethics statements
Patient consent for publication
References
Footnotes
SJ and YS contributed equally.
XZ and J-qY contributed equally.
Contributors SJ and YS contributed equally to this article in drafting and revising the manuscript. DY and PW worked on the statistical analysis for designing this trial; JX, YC and CZ drafted the informed consent form, and reviewed literature associated with the design of this trial. XZ and JY are responsible for overall supervision and instruction of the study design, are both correspondence authors for this article. JY is the guarantor.
Funding The present study is supported by the National Clinical Research Center for Cardiovascular Diseases, Fuwai Hospital, Chinese Academy of Medical Sciences (Grant No. NCRC2022003). The funder has no conflict of interest to declare and is not involved in any of the following procedures: study design, collection, analysis and interpretation of the data, writing of the report and decision to submit this paper for publication.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.