Article Text

Abstract
Introduction There are a substantial number of patients developing heart failure after transcatheter aortic valve implantation (TAVI) for severe aortic stenosis (AS), even though AS has been successfully treated. The purpose of this randomised controlled trial was to determine whether the addition of an angiotensin receptor–neprilysin inhibitor (ARNI), sacubitril/valsartan, is superior to conventional medications in lowering N-terminal pro-B-type natriuretic peptide (NT-proBNP) levels in patients undergoing TAVI for AS.
Methods and analysis The study design is a prospective, single-centre, open-label, randomised, parallel-group, two-arm study, in which participants will be randomised in a 1:1 ratio to receive either conventional medications plus ARNI or conventional medications only. In the ARNI group, if a patient was on an ACE inhibitor or angiotensin II receptor blocker before TAVI, it will be switched to ARNI 100 mg/day (50 mg two times per day) on the first postoperative day. If not, candesartan 4 mg/day will be started 1–2 days before TAVI, and switched to ARNI 100 mg/day on the first postoperative day. As the patient has tolerability to ARNI, dosage will be increased stepwise to 400 mg/day 2–4 weeks apart. ARNI will be continued until at least 6-month follow-up. In the control group, the patient will receive conventional medications. The primary endpoint is the serum NT-proBNP value at 6-month follow-up after TAVI. Each group includes 42 patients (84 total patients).
Ethics and dissemination Ethical approval for this study has been obtained from the Chiba University Hospital Certified Clinical Research Review Board (CRB3180015). The study is ongoing. Findings from this study will be disseminated through peer-reviewed publications and conference presentations.
Trial registration number This trial has been registered on the Japan Registry of Clinical Trials: jRCT1031220344.
- Adult cardiology
- Valvular heart disease
- Heart failure
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
This is a randomised controlled trial to investigate the efficacy of angiotensin receptor–neprilysin inhibitor (ARNI) in patients undergoing transcatheter aortic valve implantation for severe aortic stenosis.
The primary endpoint is the N-terminal pro-B-type natriuretic peptide value at 6-month follow-up, a well-established surrogate biomarker for heart failure.
This study incorporates a comprehensive set of secondary endpoints, including haemodynamic parameters, functional status and diuretic use, to support interpretation of the primary endpoint.
Potential limitations of the trial are that the primary endpoint is a surrogate marker for heart failure, and clinical outcomes or functional outcome measures cannot be assessed.
If this study shows the potential of ARNI for the prevention of heart failure development, it can serve as a platform for further evaluation of its prognostic impact in a larger population.
Introduction
Since previous pivotal trials have shown that transcatheter aortic valve implantation (TAVI) has a similar or better short-term and long-term prognosis compared with surgical aortic valve replacement for aortic stenosis (AS),1 2 the indications for TAVI have expanded, with the number of cases increasing in recent years. On the other hand, many studies investigating clinical outcomes after TAVI have reported that the most common cause of rehospitalisation is heart failure, with incidence ranging from 12.8% to 24.1%.3–6 In some cases, functional problems with the TAVI valve, such as severe patient–prosthesis mismatch and paravalvular leak, are known to cause heart failure after TAVI.5 7 However, most of these patients develop heart failure despite no obvious problem with TAVI valve function and preserved left ventricular ejection fraction (LVEF) on echocardiography, known as ‘heart failure with preserved ejection fraction (HFpEF)’, mainly due to left ventricular diastolic dysfunction. Even if AS is successfully treated by TAVI, HFpEF can possibly be caused by various factors, such as advanced age, residual left ventricular hypertrophy, concomitant chronic kidney disease, arrhythmia as represented by atrial fibrillation, increased afterload by hypertension and peripheral vascular resistance.3–5 Since there is no established method to prevent HFpEF after TAVI, exploring effective medications for HFpEF is necessary to improve the long-term prognosis after TAVI.
Based on the results of previous large-scale randomised controlled trials,8 administration of angiotensin receptor–neprilysin inhibitor (ARNI), sacubitril/valsartan, is recommended as one of the important options for treatment of heart failure with reduced ejection fraction (HFrEF) in the guidelines for treatment of heart failure.9 10 On the other hand, in the Prospective Comparison of ARNI with ARB (angiotensin-receptor blockers) Global Outcomes in HFpEF (PARAGON-HF) trial comparing ARNI and valsartan in patients with HFpEF, although there was no statistically significant difference in the composite primary endpoint (heart failure and cardiovascular death), ARNI was associated with a significantly lower rate of the primary endpoint in women or patients with LVEF≤57% in prespecified subgroup analyses.11 Therefore, ARNI is listed as a medication that may be considered for patients with HFpEF in the guidelines (class of recommendation is IIb).9 10 However, there are no data on the efficacy of ARNI for prevention of heart failure after TAVI. Thus, the purpose of this randomised controlled trial is to clarify whether the addition of ARNI is superior to conventional medications in lowering N-terminal pro-B-type natriuretic peptide (NT-proBNP) levels, as a diagnostic biomarker for heart failure, in patients undergoing TAVI for severe AS.
Methods
Study design
In this phase IV, prospective, single-centre, open-label, randomised, parallel-group, two-arm study, participants will be randomised in a 1:1 ratio to receive either conventional medications plus ARNI or conventional medications only.
Eligibility criteria
Eligible patients are those who meet all inclusion criteria mentioned below and none of the listed exclusion criteria. Inclusion criteria are: (1) patients with AS and symptoms of heart failure requiring TAVI based on the guidelines for management of valvular heart disease12 13; (2) patients scheduled for TAVI at Chiba University Hospital; (3) patients aged 70 years or older at the time of enrolment; and (4) patients who have provided written informed consent to participate in this study (if the patient cannot provide a signature, a family member/designate may sign on their behalf). Exclusion criteria include any of the following: (1) acute heart failure (New York Heart Association (NYHA) class IV); (2) systolic blood pressure <110 mm Hg; (3) severe hepatic dysfunction (Child-Pugh class C); (4) severe renal dysfunction (serum creatinine>3.0 mg/dL); (5) haemodialysis; (6) hyperkalaemia (serum K>5.5 mEq/L); (7) bilateral renal artery stenosis; (8) history of angioedema; (9) patient with diabetes on aliskiren fumarate; (10) a patient without written informed consent; and (11) a patient whom the investigator considers to be ineligible as a subject.
Recruitment
Recruitment for this study started in September 2022 and will end in January 2025, or until a total of 84 participants have been recruited. This study is being conducted at Chiba University Hospital.
Patient and public involvement
Patients and/or the public were not involved in the design, conduct, reporting, or dissemination plans of this research.
Sample size calculation
The target sample size for this randomised trial is 84. This number is based on feasibility and following validity. The primary analysis of this study is to test the superiority of the ARNI group (conventional medications plus ARNI) over the control group (conventional medications only) in lowering NT-proBNP levels in patients with AS undergoing TAVI. The NT-proBNP level is determined as a surrogate marker in this study, because previous studies have reported that lower NT-proBNP levels and greater reduction of NT-proBNP levels are associated with better prognosis in HFpEF patients.14 15 In the PARAGON-HF study, comparing ARNI and valsartan in patients with HFpEF, the NT-proBNP levels after 48 weeks were approximately 600 pg/mL in the ARNI group and 700 pg/mL in the control group (valsartan group).16 Since it is unlikely that the NT-proBNP level in the control group (conventional medications) in this study would be lower than that with valsartan, the NT-proBNP level after 6 months in this study was estimated to be 600 pg/mL in the ARNI group and 700 pg/mL in the control group. Assuming an SD of 150 pg/mL, an overall significance level of 5% (α=0.05), and a target power of 80% (1−β = 0.8), the minimum number of patients required is calculated to be 37 in each group (74 in total). Considering approximately 10% of discontinuation or dropout, a total sample size of 84 patients is required for this study.
Registration and assignment methods
(1) In principle, registration should be conducted within 7 days of obtaining consent. (2) The principal investigator or sub-investigators will obtain written consent and confirm that participants meet the selection criteria and do not violate the exclusion criteria as a result of the screening test. Participants deemed eligible by the principal investigator or sub-investigators will be enrolled prior to the start of study drug administration. Participants will be enrolled in the study by faxing or bringing the case registration form (CRF) with them. (3) The principal investigator and sub-investigators will complete the CRF and bring it to the data management centre at Chiba Clinical Research Center. The data centre will enter the necessary information for case registration on the data system and confirm the results of eligibility determination and assignment on the system. If the patient is found to be eligible, the investigator or sub-investigators will start the protocol treatment according to the results of the assignment. (4) Once a patient is registered, the registration will not be cancelled (deleted from the database). In case of duplicate registration, the initial registration information (registration number) will be used in all cases. In case of registration errors or duplicate registrations, the data centre shall be notified immediately.
Assignment adjustment factors
Participants will be randomly assigned in a 1:1 ratio to the study to receive conventional medications plus ARNI or conventional medications only. Eligible patients will be randomised to either the ARNI group or the control group at a ratio of 1:1, by employing a minimisation method balancing for sex (male or female), age (<85 or ≥85 years) and LVEF (<50% or ≥50%).
Study procedures
The study outline is shown in figure 1. A patient who meets the inclusion criteria and has provided written informed consent to participate in this study at least 2 days prior to the TAVI procedure will be randomly assigned to the ARNI group or the control group. In the ARNI group, ARNI will be initiated as follows, because ARNI must be administered by switching from ACE inhibitors (ACE-I) or ARB for adult heart failure based on the labelling information for prescription medicines by the Pharmaceuticals and Medical Devices Agency in Japan. If a patient is on ACE-I preoperatively, ACE-I will be stopped 1 day prior to TAVI, and ARNI 100 mg daily (50 mg two times per day) will be started on the first postoperative day. If a patient is taking an ARB preoperatively, ARB will be switched to ARNI 100 mg daily (50 mg two times per day) on the first postoperative day. If a patient is not taking ACE-I or ARB preoperatively, candesartan 4 mg/day will be started 1–2 days before TAVI, and candesartan will be switched to ARNI 100 mg daily (50 mg two times per day) on the first postoperative day. Due to the institutional restrictions on drug use, candesartan was chosen as the starter for the switch to ARNI. In a patient with tolerabiling ARNI, dosage will be increased stepwise to 400 mg/day (200 mg two times per day) 2–4 weeks apart. Tolerability is evaluated by a systolic blood pressure of 100 mm Hg or higher and a serum potassium level of <5.5 mEq/L. ARNI will be continued at least until 6-month follow-up. In the control group, patients will receive conventional medications without ARNI.

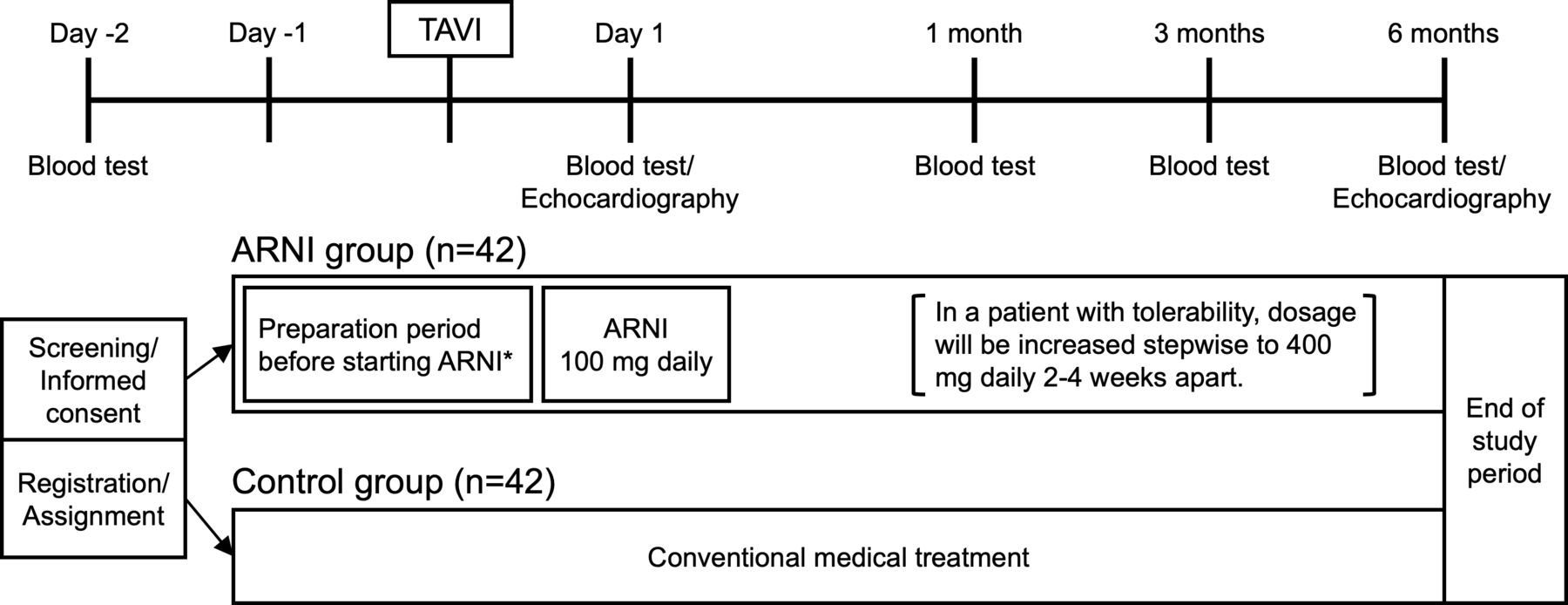{kind=link}
Graphic outline of study design. Patients are randomly assigned to either the ARNI group with conventional medications plus ARNI or the control group with conventional medications only. *ARNI will be initiated as follows, because ARNI must be administered by switching from ACE-I or ARB based on the labelling information for prescription medicines by the PMDA in Japan. (1) If a patient is on ACE-I preoperatively, ACE-I will be stopped 1 day prior to TAVI, and ARNI 100 mg daily will be started on the first postoperative day. (2) If a patient is taking an ARB preoperatively, ARB will be switched to ARNI 100 mg daily on the first postoperative day. (3) If a patient is not taking ACE-I or ARB preoperatively, candesartan 4 mg/day will be started 1–2 days before TAVI, and candesartan will be switched to ARNI 100 mg daily on the first postoperative day. ACE-I, angiotensinACE inhibitors; ARB, angiotensin II receptor blockers; ARNI, angiotensin receptor–neprilysin inhibitor; PMDA, Pharmaceuticals and Medical Devices Agency; TAVI, transcatheter aortic valve implantation.
Blinds
Blinds are not performed.
Endpoint
The primary endpoint of the study is the serum NT-proBNP value at 6-month follow-up after TAVI. The secondary efficacy endpoints include: (1) change in NT-proBNP value at 6-month follow-up; (2) systolic pulmonary artery pressure at 6-month follow-up; (3) oedema scale at 6-month follow-up; (4) NYHA class at 6-month follow-up; (5) increase or decrease in the dose of diuretics at 6-month follow-up; (6) change in body weight at 6-month follow-up; and (7) cardiovascular events (death, admission for heart failure, myocardial infarction). The secondary safety endpoints include: (1) incidence of adverse events; (2) prevalence of patients with serum K>5.5 mEq/L; and (3) prevalence of patients with symptomatic hypotension.
Statistical analysis
The full analysis set (FAS), per protocol set (PPS), and safety population will be analysed; the FAS will be the primary analysis population and all three target populations will be analysed. The distribution of subject background data and summary statistics for each analysis population will be calculated for each group. For nominal variables, category frequencies and proportions will be presented for each group. For continuous variables, summary statistics (number of cases, mean, SD, minimum, median, maximum) will be calculated for each group. For comparisons between groups, Pearson’s χ2 test is used for nominal variables, with the exception of Fisher’s direct probability calculation method for cells with an expected frequency of <5 and >20%, Wilcoxon’s rank sum test for ordinal variables and the t-test with no correspondence for continuous variables. The significance level is set at 5% (two tailed).
For the primary endpoint, the main analysis will be performed for FAS, taking the mean value in each group for NT-proBNP levels at 6 months and testing for statistical significance using an unpaired t-test. For this analysis, a similar analysis will be performed for PPS as a supplementary analysis. The significance level for the hypothesis test will be 5% (two sided). For the secondary efficacy endpoints, the analyses will be performed to provide additional insight into the primary analyses. No adjustment for multiplicity will be made in the analysis of the secondary efficacy endpoints. The significance level for hypothesis testing is 5% (two sided), and CIs are calculated as two-sided 95% CIs. For the secondary safety endpoints, which are the frequency of adverse events, tables will be prepared for the endpoints. The two-sided 95% CIs of the binomial distribution will be calculated for each group to estimate proportions. Fisher’s direct probability calculation method will be used to compare between groups if necessary.
Quality control
The monitoring manager is responsible for ensuring that the human rights of the subjects are protected, the study is conducted in accordance with the protocol, and the data are accurately collected. Central monitoring will be conducted regularly based on data from case report forms obtained at the data centre. The monitoring manager shall prepare a report that includes a summary of important findings such as diseases, nonconformities or other facts, and shall report the results of such monitoring to the principal investigator. The independent data monitoring committee, which consists of at least three expert members independent of the study, will conduct safety monitoring, including comparison of the incidence of adverse events between the study treatment and control group, and detailed examination of serious adverse events, as necessary, to ensure patient safety. The principal investigator will decide to discontinue the study as a whole if it is considered challenging to continue the study due to unforeseen adverse events, illness or other factors.
Informed consent
All participants will receive adequate information about the nature, purpose, possible risks and benefits of the trial, and on alternative therapeutic choices using informed consent approved by the IRB (online supplemental file 1). A participant must be given ample time and opportunity to ask questions and consider participating in the trial. A completed informed consent is required for enrolment in the trial. The investigators must maintain the original signed consent form and a copy of the signed consent form. To assure patient confidentiality, trial participants will be allocated a unique trial identification number for reference throughout the trial.
Supplemental material
Ethics and dissemination
This study will be conducted in accordance with the Declaration of Helsinki (revised 2013). Ethical approval for this study has been obtained from the Chiba University Hospital Certified Clinical Research Review Board (CRB3180015) and the study is registered in the Japan Clinical Trials Registry (jRCT1031220344). The investigators will explain the concept of this study to patients and obtain written informed consent from all participating patients. The initial version of the protocol was approved on 23 August 2022, and patient recruitment began on 22 September 2022. The latest version 1.5 was approved on 17 September 2024.
A manuscript summarising the results of the primary endpoint will be published in a peer-reviewed journal. Separate manuscripts for the secondary objectives will also be written and submitted for publication in peer-reviewed journals. The findings of this study will be disseminated through presentations at academic conferences.
Discussion
Although TAVI is quite an effective treatment for AS, a substantial number of patients develop heart failure after TAVI, and furthermore, heart failure after TAVI has been associated with poor prognosis.3–6 Since patients who require TAVI are often elderly and have a variety of comorbidities, they are considered to be a population at high risk of developing HFpEF even after AS is treated.17 To reduce the risk of heart failure after TAVI, comprehensive treatment that includes not only relieving AS but also managing comorbidities and administering appropriate medications may be necessary.18 19 In addition, previous studies have indicated that cardiac fibrosis and hypertrophy persist after relief of AS, leading to poor prognosis in patients with severe AS.20 Therefore, medications with an effect to inhibit cardiac fibrosis may improve prognosis after TAVI. Recently, the usefulness of sodium–glucose cotransporter 2 inhibitors (SGLT2-I) for heart failure, including HFpEF, has been reported, and thus, its use is strongly recommended in the guidelines.10 21 Although SGLT2-I may be effective in preventing heart failure after TAVI, some patients possibly have difficulty initiating SGLT2-I because of the elderly population with high frailty and risk for infections, such as urinary tract infections.22
While ARNI is widely used for the treatment of heart failure, mainly HFrEF, it is also effective to some extent for heart failure with mildly reduced ejection fraction and HFpEF.11 Previous animal studies have shown that ARNI is effective in ameliorating cardiac maladaptive remodelling, as indicated by improvements in cardiac function and decreases in cardiac fibrosis, hypertrophy and inflammation, suggesting that ARNI may also be beneficial after TAVI.23 24 However, it has been unclear whether ARNI can actually reduce the risk of heart failure after TAVI. Therefore, this randomised study has been designed to investigate this issue. This study is designed to determine the impact of adding ARNI to conventional medications, such as antihypertensive agents and diuretics. Although most of the past studies showing the efficacy of ARNI in heart failure have compared it with ACE-I or ARB,8 11 this study was designed to compare conventional medications plus ARNI with conventional medications only, because the efficacy of ACE-I or ARB in heart failure after TAVI has not been demonstrated. Since NT-proBNP has been used as an important parameter for the evaluation of heart failure in many previous studies examining the efficacy of ARNI,16 NT-proBNP level at follow-up is set as the primary endpoint in this study. If this study shows the usefulness of ARNI for the prevention of heart failure, the next step may be to investigate whether it can reduce heart failure events in a larger-scale study. In such a future study, functional outcome measures, for example, a 6 min walk test, which is not planned to investigate in this study, should also be examined.
Ethics statements
Patient consent for publication
Acknowledgments
We thank the Translational Research and Development Centre, Chiba University Hospital, for their cooperation in protocol development. We thank Heidi N Bonneau, RN, MS, CCA for her editorial review of the manuscript.
References
Footnotes
Contributors HK designed the original concept. All coauthors contributed significantly to the conception and design of the study, with specific additional contributions from each coauthor within their area of expertise. The protocol was written by HK, SO and KM, and it was critically reviewed by TS, HYag, HG, HYam, TK, YI, HH, GM and YK. All authors gave approval for the publication. YK is the guarantor.
Funding This study is funded by a CREATION-Fund (of Chiba University Hospital) and Clinical Research Initiation-Fund (of Chiba University Hospital).
Competing interests HK, SO, HYag, HG, HYam, TK, KM, GM and YK are affiliated with Chiba University Graduate School of Medicine, to which Chiba University Hospital belongs, and ST, IY and HH are affiliated with Chiba University Hospital, which provided financial support for this study. However, the institutions were not involved in the design, execution, data analysis or interpretation of the results in this study. YK received lecture fees from Novartis Pharma
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.