Article Text

Abstract
Objectives To investigate the long-term safety and efficacy of otilimab, an antigranulocyte-macrophage colony-stimulating factor monoclonal antibody, for patients with rheumatoid arthritis (RA).
Methods ContRAst X (NCT04333147) was a phase 3, multicentre, long-term extension trial. Patients with RA aged ≥18 years who completed a qualifying contRAst trial (contRAst 1–3) and who the investigator thought might benefit from long-term otilimab treatment were eligible to enter contRAst X. Patients who received otilimab (90 mg/150 mg) in their qualifying trial maintained the same dose; patients who received tofacitinib or sarilumab were rerandomised 1:1 to either otilimab dose. Patients could continue background conventional synthetic disease-modifying antirheumatic drugs. The primary objective was long-term safety (up to 4 years).
Results Of the 2916 patients who entered contRAst X, 2915 received otilimab (exposure range: 7–896 days); the majority were withdrawn due to early trial termination. For otilimab 90 mg and 150 mg, the incidence of adverse events (AEs) was 62% (n=902/1456) and 64% (n=931/1459), the incidence of AEs of special interest was 8% (n=120/1456) and 7% (n=95/1459) and the incidence of serious AEs was 8% (n=123/1456) and 8% (n=114/1459), respectively. There were no instances of pulmonary alveolar proteinosis (PAP), active tuberculosis (TB), TB reactivation or serious hypersensitivity reactions. The proportions of clinical disease activity index low disease activity responders remained relatively stable throughout, with no apparent reduction following the switch from tofacitinib/sarilumab to otilimab.
Conclusion No new safety signals or instances of PAP were associated with long-term (≤2.5 years) treatment with otilimab.
Trial registration number ClinicalTrials.gov: NCT04333147.
- RHEUMATOLOGY
- THERAPEUTICS
- Clinical Trial
Data availability statement
Data are available upon reasonable request. GSK has a long-standing commitment to clinical data transparency. The GSK Study Register is an online portal where anyone can access information about the clinical research GSK carries out on its existing vaccines and medicines. GSK registers protocol summaries before the start of a trial and posts results summaries within a year of study completion. Since 2020, GSK has developed plain language summaries of the results of our phase 2–4 clinical trials, which are shared with the study investigator for distribution to trial participants and are available on the GSK Study Register and www.trialsummaries.com. GSK is committed to providing access to anonymised patient-level data that sit behind the results of clinical trials. GSK-sponsored interventional clinical trials will be listed for data sharing once a medicine has been approved by regulators or terminated from development, and the study has been accepted for publication. External researchers can request access to anonymised patient-level clinical trial data and supporting clinical trial documents through the multisponsor data-sharing portals. Further details are available at: http://www.gsk.com/en-gb/innovation/trials/data-transparency/ and http://www.gsk-studyregister.com/en/.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
This long-term extension study of the antigranulocyte-macrophage colony-stimulating monoclonal antibody, otilimab, included a large, global population of almost 3000 patients with rheumatoid arthritis.
The trial population comprised subgroups of patients with varying refractory status: from those with an inadequate response to methotrexate only, to those ‘difficult-to-treat’ patients with a prior inadequate response to conventional synthetic and biologic disease-modifying antirheumatic drugs and/or Janus kinase inhibitors.
Another strength of the methods was the intended trial duration of approximately 4 years which would have provided extensive data on the long-term safety of otilimab if the trial had not been terminated early.
Due to the theoretical risk of pulmonary alveolar proteinosis and to align with the dosing strategy used in the qualifying trials, a higher otilimab dose was not permitted, reflecting a potential limitation of the study.
Introduction
Preclinical evidence has suggested a possible role of granulocyte-macrophage colony-stimulating factor (GM-CSF) in the pathogenesis of rheumatoid arthritis (RA) and the mediation of pain,1–8 and consequently, the GM-CSF pathway was considered an attractive target for the development of new biologic therapies for the treatment of RA, as discussed previously.9 10 Otilimab, a high-affinity anti-GM-CSF monoclonal antibody,11 was investigated for the treatment of patients with active RA in the multicentre phase 3 contRAst programme.9 10 The contRAst programme comprised three double-blind randomised controlled trials (contRAst 1–3) and one long-term extension (LTE) trial (contRAst X) to investigate the safety and efficacy of otilimab over an extended period.
ContRAst 1 and contRAst 2 were 52-week trials in patients with a prior inadequate response to either methotrexate (MTX; contRAst 1) or conventional synthetic/biologic disease-modifying antirheumatic drugs (cs/bDMARDs; contRAst 2). ContRAst 3 was a 24-week trial that included patients with an inadequate response to csDMARDs and bDMARDs and/or Janus kinase (JAK) inhibitors.9 10 All three qualifying trials compared weekly subcutaneous (SC) injections of otilimab 90 mg and otilimab 150 mg with placebo and with the active comparators, tofacitinib (5 mg orally two times per day in contRAst 1 and contRAst 2) and sarilumab (200 mg SC every 2 weeks in contRAst 3) in combination with background MTX (contRAst 1) or csDMARDs (contRAst 2 and contRAst 3).9 10 Tofacitinib and sarilumab were considered appropriate comparators, given the EU (European Union) and US recommendations to switch to a bDMARD or targeted synthetic DMARD of a different class, such as an interleukin-6 or JAK inhibitor, following the failure of a tumour necrosis factor inhibitor.12 13 After week 12, patients receiving placebo were switched to one of the active treatments for the remainder of the trial. Following the completion of the qualifying trial, eligible patients had the option to enter contRAst X.9 10 The primary endpoint in the three qualifying trials was the proportion of patients achieving an American College of Rheumatology 20 (ACR20) response, indicated by at least a 20% improvement in the ACR core set measures, at week 12 for otilimab versus placebo.9 10 14 ContRAst 1 and contRAst 2 met the primary endpoint. It was also observed that a greater proportion of patients treated with otilimab achieved Clinical Disease Activity Index (CDAI) low disease activity (LDA) and improved Health Assessment Questionnaire-Disability Index (HAQ-DI) scores compared with placebo; however, otilimab was inferior to tofacitinib across multiple endpoints.9 In contRAst 3, although numerically more patients were ACR20 responders with either otilimab dose versus placebo, this was not statistically significant, and thus, the trial failed to meet the primary endpoint. As with contRAst 1 and contRAst 2, otilimab was consistently inferior to the active comparator (sarilumab) across multiple endpoints in contRAst 3.10 In all three trials, within the reported time frame, a similar incidence of adverse events (AEs) was noted with otilimab and the approved active comparators.9 10
Here we report the results of contRAst X, which investigated the long-term safety of otilimab in combination with csDMARDs, as well as the ability of otilimab to maintain efficacy in patients who achieved an initial clinical response in a qualifying contRAst trial.
Methods
Trial design
ContRAst X (study number 209564; NCT04333147) was a phase 3, multicentre, LTE trial conducted at 387 sites across 27 countries (online supplemental table 1). Enrolment began on 12 May 2020, and the intended trial duration was approximately 4 years to enable patients to receive otilimab until it was anticipated to become commercially available. However, after pivotal results were obtained in the qualifying trials, the LTE was terminated early on 22 October 2022, and discontinuation of the contRAst programme was announced to the public on 27 October 2022.15 The last patient dose occurred on 16 December 2022, and the last safety follow-up occurred on 28 February 2023.
Supplemental material
Otilimab was administered SC by prefilled syringe in most patients and by autoinjector in 184 patients. Patients who received otilimab 90 mg or 150 mg in their qualifying trial continued to receive the same dose in contRAst X, whereas those who received tofacitinib or sarilumab were rerandomised 1:1 to receive either otilimab dose (online supplemental figure 1).
The time points reported in this manuscript refer to the number of weeks since enrolment in contRAst X. Therefore, patients who entered contRAst X from an otilimab arm in the qualifying trial received otilimab for the number of weeks reported, and an additional 40–52 weeks (if entering from contRAst 1 and contRAst 2) or an additional 12–24 weeks (if entering from contRast 3). For safety assessments, ‘baseline’ was defined as the assessment before the patient’s first dose of otilimab (administered either in contRAst X or in the qualifying trial for those entering contRAst X from an otilimab arm of the qualifying trial). For efficacy parameters, ‘baseline’ was defined as the assessment before the first dose of otilimab administered in contRAst X.
The trial was conducted in accordance with the Declaration of Helsinki, International Conference on Harmonisation, Good Clinical Practice and applicable country-specific regulatory requirements. The protocol was approved by the Health Research Authority and Health and Care Research Wales (Research Ethics Committee reference: 20/SC/0172) and the relevant Institutional Review Board/Independent Ethics Committee for each individual site (online supplemental table 1; reference numbers not available). All patients provided written informed consent.
Patients
Eligible patients were male or female adults (aged ≥18 years) with RA who completed one of the qualifying contRAst trials and, in the investigator’s judgement, may have benefitted from long-term treatment with otilimab. Patients were excluded if the trial intervention was permanently discontinued at any time during the qualifying trial or temporarily discontinued at the time of the final visit in the qualifying trial. Full eligibility criteria are provided in the online supplemental materials.
Supplemental material
Randomisation and blinding
Rerandomisation of patients switching from tofacitinib or sarilumab to otilimab was conducted using interactive voice response technology. Patients’ treatment allocation remained blinded at least until their qualifying trial had been reported. Patients could continue in contRAst X if their intervention assignment was unblinded.
Concomitant medications
Background csDMARDs (two or less) from the qualifying trials could be maintained. If clinically indicated, dose reduction or discontinuation was permitted after the completion of week 12 of contRAst X for patients who joined from contRAst 1 and contRAst 2 or after week 24 for those who joined from contRAst 3. See online supplemental materials for permitted and prohibited medications.
Endpoints and assessments
The primary objective was the long-term safety of otilimab at weekly doses of 90 mg or 150 mg; primary endpoints included incidence of AEs, serious AEs (SAEs), AEs of special interest (AESI) and other important AEs. An AE was considered trial intervention-emergent if the AE onset date was on or after the intervention start date in contRAst X and on or before the safety follow-up visit (8 weeks postlast dose). AESIs for otilimab included serious infections, opportunistic infections, latent tuberculosis (TB), active TB, TB reactivation, grade ≥3 neutropenia (<1.0×109 neutrophils/L), pulmonary alveolar proteinosis (PAP), serious hypersensitivity reactions, injection site reactions, persistent grade ≥2 cough per Common Terminology Criteria for Adverse Events (CTCAE) and persistent grade ≥2 dyspnoea per dyspnoea scale. Other important AEs were protocol-defined, adjudicated cardiovascular (CV) events, adjudicated gastrointestinal (GI) perforation, malignancy, herpes infection, all-cause mortality, serious pulmonary infections, pneumonia (serious and non-serious), hepatitis B virus (HBV), HBV reactivation and thromboembolic events. Additional safety assessments included worst-grade shift from baseline in haematology and clinical chemistry parameters (graded by National Cancer Institute (NCI)-CTCAE) and the proportion of patients with NCI-CTCAE ≥grade 3 haematological/clinical chemistry abnormalities.
Long-term efficacy was a secondary objective. Disease activity states were defined using the standard CDAI cut-off values for high (>22), moderate (>10 to ≤22) and low (≤10) disease activity and remission (≤2.8). Major secondary endpoints were assessed at weeks 12 and 24 and every 48 weeks thereafter and included the proportion of patients with CDAI LDA and in CDAI remission (CDAI high and moderate disease activity were assessed as posthoc analyses); ACR/EULAR/EULAR remission (Boolean and simplified disease activity index (SDAI)); absolute values of CDAI total score, HAQ-DI, Pain Visual Analogue Scale (VAS), Short Form-36 (SF-36) Physical Component Summary (PCS) and Mental Component Summary (MCS) scores, Functional Assessment of Chronic Illness Therapy (FACIT)-Fatigue and immunogenicity. See online supplemental table 2 for a full list of outcome measures.
Statistical analysis
There were no formal statistical hypotheses or sample size calculations. Sample size was ultimately determined by the number of patients who, in the investigator’s judgement, might benefit from long-term otilimab treatment and who consequently elected to enter contRAst X. The primary analysis population was the safety population, defined as all enrolled patients who received ≥1 dose of trial intervention; these patients were analysed according to the intervention they received, and data were summarised descriptively. Efficacy was analysed in the intent-to-treat (ITT) population, defined as all enrolled patients who received ≥1 dose of trial intervention; these patients were analysed according to the planned treatment. Posthoc analyses of subgroups stratified by the prior treatment arm in the qualifying trial were performed for patient demographics, baseline characteristics and selected safety and efficacy endpoints.
Patient and public involvement
Patients were involved in patient advisory boards and in-person touchpoints in which the trial design and endpoints were discussed. There was no further patient or public involvement in the conduct or reporting of the trial.
Results
Trial population
The majority of the 3148 patients who completed a qualifying trial enrolled in contRAst X (N=2916; 92.6%), of whom 1169 entered from contRAst 1, 1282 from contRAst 2 and 465 from contRAst 3. Of the 2916 enrolled patients, 2915 received open-label otilimab and were included in the safety and ITT populations (figure 1). Following the completion of contRAst 1 and contRAst 2, a similar proportion of patients initially randomised to the otilimab 90 mg, otilimab 150 mg or tofacitinib arms enrolled in contRAst X. In contRAst 1, 75% and 76% of patients enrolled from the otilimab 90 mg and 150 mg arms, respectively, and 79% enrolled from the tofacitinib arm. In contRAst 2, 75% enrolled from each of the otilimab arms and 77% enrolled from the tofacitinib arm. In contRAst 3, compared with sarilumab, a slightly higher proportion of patients initially randomised to the otilimab 90 mg or 150 mg arms enrolled in contRAst X (otilimab 90 mg: 85%; otilimab 150 mg: 89%; sarilumab: 80%). Most patients were withdrawn from contRAst X due to early termination of the trial.

Patient disposition. Figure indicates the number of patients who enrolled in contRAst X from each of the qualifying trials (contRAst 1, contRAst 2 or contRAst 3) and the total number enrolled. Green boxes indicate the treatment arm of the qualifying trial that patients were enrolled on. Blue boxes indicate the safety and intent-to-treat (ITT) populations of contRAst X, stratified by prior treatment in the qualifying trial. *Patients who received otilimab in their qualifying trial were assigned to the same dose in contRAst X. †Patients who received an active comparator (tofacitinib or sarilumab) were rerandomised 1:1 to either otilimab dose. ‡One patient was randomised to the otilimab 150 mg arm but was withdrawn due to a physician decision before receiving any dose, and this patient was excluded from the safety and ITT populations. §One death due to septic shock occurred after the safety follow-up visit (starting 64 days and resulting in death 65 days after the last dose) and thus was not considered treatment emergent.
On the date of trial termination, 2602 (89%) of the enrolled patients were remaining in the trial; the range of exposure to otilimab from the first dose in contRAst X up to and including the last patient dose was 7–889 days for otilimab 90 mg, 7–896 days for otilimab 150 mg (7–896 days overall); the total treatment exposure was 1317.7 person-years in the otilimab 90 mg arm and 1326.5 person-years in the otilimab 150 mg arm (note: exposure values do account for breaks in treatment/missed doses).
At weeks 12, 48 and 84, the proportions of patients remaining in the trial were 98%, 55% and 20%, respectively. Due to the declining number of patients, leading to increased variability in efficacy parameters after 84 weeks, efficacy data are only presented up to week 84.
Posthoc analyses of baseline demographics and clinical characteristics are reported for subgroups of patients stratified by the treatment they received in their qualifying trial and were generally balanced (table 1).
Baseline* demographics and clinical characteristics in all patients, stratified by treatment arm of the qualifying trial
Primary endpoint: safety
All safety data reported are events that emerged during contRAst X only; similar results were observed with both otilimab doses. The incidence of AEs in the otilimab 90 mg and otilimab 150 mg arm was 62% and 64%, respectively (table 2). The incidence of each AESI was similar between the two otilimab arms; serious infections occurred in 2% of patients in either arm, as did injection site reactions. Despite the 3% and 2% incidence of latent TB in the otilimab 90 mg and otilimab 150 mg arms, respectively, there were no events of TB reactivation or active TB. Opportunistic infections, neutropaenia and persistent cough occurred in <1% of patients in either otilimab arm, and there were no reports of PAP or serious hypersensitivity reactions with either otilimab dose (table 2). The incidence of other important AEs, including adjudicated CV events, adjudicated GI perforation and any malignancy, was <1% in each otilimab arm. The incidence of any herpes infection was 2% in each arm, whereas that of thromboembolic events was <1% in the otilimab 90 mg arm and 1% in the otilimab 150 mg arm (table 3). The incidence of SAEs was 8% in each otilimab arm (online supplemental table 3). There were 10 (<1%) fatal SAEs in the otilimab 90 mg arm, one of which (adenocarcinoma of pancreas) was considered drug-related by the investigator; and 9 (<1%) fatal SAEs in the otilimab 150 mg arm (online supplemental table 4), none of which were considered drug-related by the investigator.
Summary of treatment-emergent AEs and AESI incidence
Summary of other important treatment-emergent AE incidence
The proportion of patients who had a worst-grade shift to grade 3 or 4 from baseline in key laboratory parameters was low (≤1%; online supplemental table 5). Decreased lymphocyte count was most common, with a worst-grade shift to grade 3 occurring in 1% of patients in each group and to grade 4 in <1% of patients in the otilimab 150 mg group only. A worst-grade shift to either grade 3 or 4 in decreased neutrophil count occurred in <1% of patients in both groups (online supplemental table 5). Patients who entered contRAst X from the sarilumab arm of contRAst 3 had lower baseline neutrophil and platelet counts than those who entered from an otilimab arm (online supplemental table 6). After an initial worsening at week 1 in the prior sarilumab subgroups, gradual improvements in both counts were observed up to week 12, with no further changes observed with prolonged exposure to otilimab, and no changes in the other subgroups (online supplemental figure 2).
Secondary endpoints: efficacy
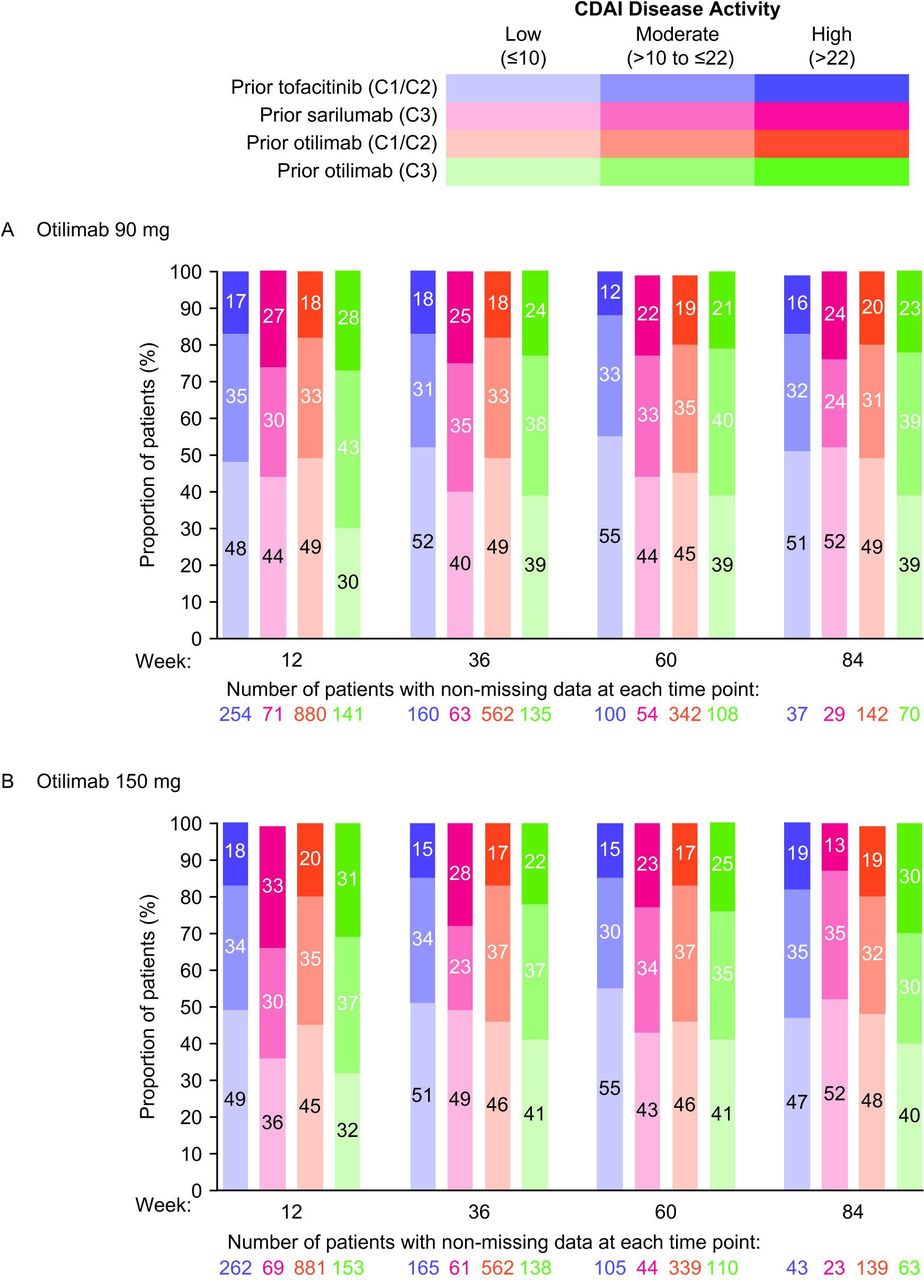
Between weeks 12 and 84, the proportions of patients with CDAI LDA (CDAI≤10) remained relatively stable and ranged from 44.4%–48.4% for otilimab 90 mg and 43.9%–46.7% for otilimab 150 mg (online supplemental figure 3A). Posthoc analysis of patient subgroups stratified by treatment arm in the qualifying trial did not appear to demonstrate reductions in CDAI LDA responders over time or increases in the number of patients with moderate or high CDAI disease activity over time, following the switch from tofacitinib or sarilumab to otilimab at week 0 of contRAst X (figure 2). However, marginal increases from baseline in CDAI total scores were observed in patients who switched from tofacitinib at week 0 (online supplemental figure 3B,C). Nevertheless, the proportions of patients with ≥50% reductions from baseline in CDAI total score increased over time in each of the prior treatment subgroups except in the prior tofacitinib subgroup of the otilimab 90 mg arm, where proportions decreased or remained the same. However, it should be noted that the proportions of patients with ≥50% reductions from baseline in CDAI total score were already low at week 12 (approximately 15%) and increased to only approximately 25% at week 84, which could have also been due to the declining patient numbers during this LTE study (online supplemental figure 4). The proportion of patients achieving a minimally important reduction from baseline (≥6) in CDAI total score also increased over time for all subgroups receiving otilimab 90 mg and for the contRAst 3 subgroups receiving otilimab 150 mg (online supplemental figure 5A,B).

{kind=link}
{kind=link}
Proportion of patients with CDAI low, moderate and high disease activity with (A) otilimab 90 mg and (B) otilimab 150 mg (posthoc analysis). C1, contRAst 1; C2, contRAst 2; C3, contRAst 3; CDAI, Clinical Disease Activity Index.
In patients who were not CDAI LDA responders, increases from baseline in CDAI total score were observed in the majority of subgroups at each time point (online supplemental figure 6). The proportion of CDAI LDA non-responders who failed to achieve minimum important reductions from baseline in CDAI total scores ranged from 64%–91% across subgroups over time (online supplemental figure 5C,D). On the other hand, CDAI LDA responders demonstrated reductions from baseline in CDAI total score between weeks 12 and 84. In particular, the subgroup of patients who entered contRAst X from both otilimab arms of contRAst 3 appeared to demonstrate greater reductions from baseline in CDAI total scores than patients in any other subgroup (online supplemental figure 6).
Between Weeks 12 and 84, the proportions of patients in CDAI remission (CDAI total score ≤2.8) remained relatively stable at approximately 10%–11% for otilimab 90 mg and ranged from 8%–11% for otilimab 150 mg (online supplemental figure 7A). The proportions of patients in SDAI remission ranged from 10%–12% in the otilimab 90 mg group and from 8%–10% in the 150 mg group, whereas the proportions of patients in Boolean remission ranged from 6%–8% and from 4%–7%, respectively (online supplemental figure 7B,C).
Mean HAQ-DI scores at week 84 were 1.08 and 1.06 with otilimab 90 mg and 150 mg, respectively, and had remained relatively stable and similar between the two doses at each time point (online supplemental figure 8A). In the subgroups of patients stratified by the prior treatment arm in the qualifying trial, no trends were observed in change from baseline in HAQ-DI or in the proportions of patients reporting minimally clinically important differences (MCID: ≥0.22 reduction from baseline; posthoc analysis data not shown). Similarly, mean pain VAS scores between weeks 12 and 84 remained relatively stable at 35–38 with either otilimab dose (online supplemental figure 8B). There were no changes in SF-36 PCS, MCS or FACIT-Fatigue scores over time with prolonged exposures to otilimab (online supplemental figure 9A–C).
Otilimab serum concentrations were similar to those observed in the qualifying trials and were sustained until the last measurement at week 48. As previously observed, otilimab 150 mg resulted in higher serum concentrations than otilimab 90 mg (data not shown). Most patients tested negative for antidrug antibodies throughout the trial (93% and 94% for otilimab 90 mg and 150 mg, respectively).
Discussion
In this LTE trial, no new safety signals were identified with prolonged otilimab treatment compared with the qualifying trials of up to 1 year duration. There were no differences in safety profiles between otilimab 90 mg and 150 mg, with the incidence of AEs, SAEs and individual AESIs generally balanced across both doses and similar to those observed in the qualifying trials.9 10
Given the crucial role of GM-CSF in the clearance of lung surfactant and debris by alveolar macrophages, there have previously been concerns that the inhibition of GM-CSF with a therapeutic monoclonal antibody could result in the development of PAP. It has also been suggested that risk factors present in the RA patient population, such as sex, smoking status and serology, may predispose patients to developing rheumatoid-related lung disease and potentially PAP.16 Due to these concerns, a maximum otilimab dose was mandated by regulators during the early phase of development. Additionally, close monitoring of pulmonary assessments and lung biomarkers associated with PAP, as well as review by a pulmonary adjudication panel, where relevant, has been a key aspect of phase 2 and phase 3 otilimab trials.9 10 16 The patient population included in the contRAst programme was large and broad enough to encompass those patients who may have an increased risk of developing PAP,17 and throughout the three qualifying trials and the LTE, no evidence of PAP was reported.9 10 This is consistent with previous trials of otilimab and other anti-GM-CSF therapies and suggests that, to date, the risk remains primarily theoretical.6 18 19 One potential explanation for the lack of evidence of PAP could be that, rather than complete neutralisation, the monoclonal antibody may in fact redistribute the cytokine from the inflammatory site only, thereby leaving basal GM-CSF levels in the lung largely unimpacted, as previously proposed by Piccoli et al.20 Another explanation may be that the safety concerns were originally identified based on observations from murine studies that did not translate into clinical findings, and thus highlight a limitation of using murine models to guide dosing in clinical trials.
Another potential safety risk associated with GM-CSF inhibition is the impact on immunological responses due to downstream inhibition of tumour necrosis factor-α production leading to an increased risk of infection such as TB.21 22 Importantly, despite the presence of latent TB in 2%–3% of patients, there were no events of active TB or TB reactivation with prolonged exposure to otilimab. Furthermore, there were no serious hypersensitivity reactions with prolonged exposure to otilimab.
Efficacy analyses demonstrated that prolonged treatment with otilimab maintained the clinical responses achieved with otilimab or the active comparators in the qualifying trials, as the proportion of CDAI responders did not appear to decline following the transition into contRAst X. In fact, in the difficult-to-treat ‘refractory’ patients who entered the LTE from contRAst 3, the CDAI total scores declined, and the proportions achieving a minimum important reduction in CDAI score increased over time. Nevertheless, although the CDAI response in contRAst X remained relatively consistent with contRAst 1–3, the efficacy of otilimab compared with the approved treatments in the qualifying trials was not sufficient to warrant further development in RA,9 10 and the decision was made by the sponsor to terminate the LTE.
It is noteworthy that although more than 50% of patients were CDAI LDA non-responders in contRAst X, the majority of patients (86%) remained in the trial until it was terminated. One potential reason for this may be that these patients experienced reasonable improvements in disease activity despite not achieving LDA. Another possible reason, especially for those patients who entered from contRAst 3, is the lack of alternative therapies for patients who had previously failed biologic and/or targeted synthetic DMARDs. It is also possible that the level of care and attention provided in a clinical trial were of value from the patient’s perspective, and patients may have chosen to remain in the trial as long as they did not feel that their condition was worsening or for fear of the unknown if they were to withdraw. Socioeconomic issues such as health insurance and affordability of targeted DMARDs may have also influenced patients’ decisions.
As noted previously, clinically meaningful improvements in endpoints such as pain VAS in the phase 2 trial, BAROQUE (Bringing Anti-GM-CSF to Rheumatoid Arthritis: A New Approach to Overcoming an InadeQUate ResponsE to MTX),18 were not replicated in the phase 3 programme.9 10 A trend of lesser response has been increasingly observed in RA clinical phase 3 trials, potentially due to factors such as broader inclusion criteria and smaller sample sizes in phase 2 trials resulting in overestimation of treatment effects.23 24 Two alternative GM-CSF–targeting molecules have previously been investigated for the treatment of RA, with mixed results, so that neither agent progressed beyond phase 2.6 9 10 18 19 Efficacy results from this phase 3 contRAst programme confirm that, despite a plausible hypothesis,6 19 targeting GM-CSF alone does not achieve adequate control of disease activity in RA.
The dosing strategy used in contRAst X was consistent with that used in the qualifying trials; a limitation of which being that an even higher otilimab dose was not permitted by the regulatory authorities due to the theoretical risk of PAP. As previously reported, the doses administered resulted in higher than predicted serum otilimab concentrations and similar efficacy between doses.9 10 The lack of an observed dose-response makes it unlikely that a higher dose would have resulted in greater efficacy, should that have been permitted.9 10
Strengths of the phase 3 programme were the global patient population, inclusion of difficult-to-treat, ‘refractory’ patients and inclusion of both placebo and active head-to-head comparators in the qualifying trials to evaluate the efficacy and safety of otilimab against approved targeted therapies.
In conclusion, in this trial of almost 3000 patients with RA treated with otilimab for up to 2.5 years, no new safety signals compared with the placebo-controlled trials nor instances of PAP were reported. Given the limited efficacy of otilimab compared with tofacitinib and sarilumab in contRAst 1–3 and the lack of superiority to placebo in contRAst 3, the decision was made to terminate this LTE trial and any further development of otilimab in RA.
Data availability statement
Data are available upon reasonable request. GSK has a long-standing commitment to clinical data transparency. The GSK Study Register is an online portal where anyone can access information about the clinical research GSK carries out on its existing vaccines and medicines. GSK registers protocol summaries before the start of a trial and posts results summaries within a year of study completion. Since 2020, GSK has developed plain language summaries of the results of our phase 2–4 clinical trials, which are shared with the study investigator for distribution to trial participants and are available on the GSK Study Register and www.trialsummaries.com. GSK is committed to providing access to anonymised patient-level data that sit behind the results of clinical trials. GSK-sponsored interventional clinical trials will be listed for data sharing once a medicine has been approved by regulators or terminated from development, and the study has been accepted for publication. External researchers can request access to anonymised patient-level clinical trial data and supporting clinical trial documents through the multisponsor data-sharing portals. Further details are available at: http://www.gsk.com/en-gb/innovation/trials/data-transparency/ and http://www.gsk-studyregister.com/en/.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants. The trial was conducted in accordance with the Declaration of Helsinki, International Conference on Harmonisation, Good Clinical Practice, and applicable country-specific regulatory requirements. The protocol was approved by the Health Research Authority and Health and Care Research Wales (Research Ethics Committee reference: 20/SC/0172) and the relevant Institutional Review Board/Independent Ethics Committee for each individual site (online supplemental table 1; reference numbers not available). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors would like to thank Katherine Davy, Mark Layton and Jatin Patel for their support in the design of the trial. Editorial support (in the form of writing assistance, including preparation of the draft manuscript under the direction and guidance of the authors, collating and incorporating authors’ comments for each draft, assembling tables and figures, grammatical editing and referencing) was provided by Katie Ryan, PhD; Clare Cunningham, PhD and Chrystelle Rasamison of Avalere Health, UK, and was funded by GSK.
References
Footnotes
Contributors RMF contributed to the conception/design of the trial and the acquisition and analysis/interpretation of data. MEW, PCT, VS, MW, DS, AG, and DB contributed to the trial conception or design and data analysis or interpretation. IBMcI, TT, RW, KR, MB, RN, CS, CO’S and PC contributed to data analysis or interpretation. TA contributed to the acquisition of data and data analysis or interpretation. All authors contributed to drafting or critically revising the article and provided final approval and agreement of accountability. DS acted as guarantor.
Funding This trial (GSK ID: 209564) was funded by GSK.
Competing interests MEW receives research support from AbbVie, Aqtual, Bristol Myers Squibb and Lilly and consultation fees from AbbVie, Aclaris, Amgen, Bayer, Bristol Myers Squibb, CorEvitas, Genosco, Gilead Sciences, GSK, Horizon, Johnson & Johnson, Lilly, Novartis, Pfizer, Rami Therapeutics, R Pharma, Roche, Sanofi, Scipher, Sci Rhom, Set Point and Tremeau. He holds stock/stock options in CanFite, Inmedix and Scipher. PCT has received consulting fees from AbbVie, AnaptysBio, Acelyrin, Biogen, Immunovant, Fresenius, Galapagos, Gilead Sciences, GSK, Janssen, Lilly, Nordic Pharma, Pfizer, Roche, Sanofi and UCB and research support from Galapagos. IBMcI has received consultancy and research support from AbbVie, Amgen, AstraZeneca, Bristol Myers Squibb, Causeway, Compugen, Gilead Sciences, GSK, Lilly, Novartis, Pfizer and UCB and holds a leadership role in the University of Glasgow, Versus Arthritis and is an NHS GGC Board Member and an Annals of the Rheumatic Diseases Editorial Board Member. TA has accepted research grants and/or honoraria for meetings from AbbVie, Alexion, Astellas Pharma, Bristol Myers Squibb, Chugai Pharmaceutical, Daiichi Sankyo, Eisai, Gilead Sciences, GSK, Lilly Japan, Mitsubishi Tanabe Pharma, Otsuka Pharmaceutical, Pfizer, Takeda Pharmaceutical and UCB Japan. VS has received consulting fees from AbbVie, Alpine, Alumis, Amgen, Aria, AstraZeneca, Bayer, Bristol Myers Squibb, Boehringer Ingelheim, Celltrion, Ermium, Genentech/Roche, GSK, Horizon, Inmedix, Janssen, Kiniksa, Lilly, Merck, MiMedx, Novartis, Omeros, Pfizer, R-Pharm, RAPT, Regeneron, Samsung, Sandoz, Sanofi, Scipher, Setpoint, Sorrento, Spherix, Tonix and Urica. TT received payment or honoraria from AbbVie, Asahi Kasei, Astellas, AstraZeneca, Bristol Myers Squibb, Chugai Pharmaceutical, Daiichi Sankyo, Eisai, Gilead Sciences, Janssen, Lilly Japan, Mitsubishi-Tanabe and Pfizer Japan and is an Annals of the Rheumatic Diseases Editorial Board Member. MB, DB, PC, AG, RN, CO’S, DS, CS, MW and RW are employees of GSK and hold GSK stock/shares. KR is an employee of Probabilitas Consulting Limited and is contracted by GSK. RMF has received research support from AbbVie, Amgen, Arthrosi, AstraZeneca, Biogen, Bristol Myers Squibb, Boehringer Ingelheim, Galvani, Genentech/Roche, Gilead, GSK, Janssen, Lilly, Novartis, Pfizer, Priovant, Samsung, Sanofi-Genzyme, Selecta and UCB; consulting fees from AbbVie, Amgen, Arthrosi, Bristol Myers Squibb, Cyxone, Dren Bio, Galapagos, Galvani, Gilead, GSK, Immunovant, ImmuneMed, InventisBio, Janssen, Kiniksa, Lilly, Monte Rosa, Novartis, Pfizer, Priovant, Recor, Samsung, Synact, UCB and VYNE and is an Annals of the Rheumatic Diseases Editorial Board Member.
Patient and public involvement Patients and/or the public were involved in the design, conduct, reporting or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.