Article Text

Abstract
Introduction Alcohol use disorder (AUD) is a massive burden for the individual, relatives and society. Despite this, the treatment gap is wide compared with other mental health disorders. Treatment options are sparse, with only three Food and Drug Administration (FDA)-approved pharmacotherapies. Glucagon-like peptide-1 (GLP-1) receptor agonists have shown promising effects in reducing alcohol consumption in preclinical experiments, and clinical trials are in high demand to investigate these potentially beneficial effects in patients diagnosed with AUD.
Methods and analysis The effects of the once-weekly GLP-1 receptor agonist semaglutide will be investigated in a 26-week, randomised, placebo-controlled, double-blinded clinical trial. 108 patients diagnosed with AUD and comorbid obesity (body mass index (BMI)≥30 kg/m2)) will be randomised to treatment with either semaglutide or placebo in combination with cognitive behavioural therapy. A subgroup of the patients will have structural, functional and neurochemical brain imaging performed at baseline and after 26 weeks of treatment. The primary endpoint is the reduction in heavy drinking days, defined as days with excess consumption of 48/60 g of alcohol per day (women and men, respectively). Secondary endpoints include changes from baseline to week 26 in alcohol consumption, smoking status, quality of life, fibrosis-4 score, plasma concentration of phosphatidylethanol, brain gamma-aminobutyric acid (GABA) levels, alcohol cue reactivity, functional connectivity and white matter tract integrity.
Status Recruitment started in June 2023.
Ethics and dissemination The study is approved by the Ethics Committee of the Capital Region of Denmark, the Danish Board of Health and the Danish Data Protection Agency. All patients will sign the written consent form before being included in the trial. Results will be disseminated through peer-reviewed publications and conference presentations. After the results are published, all de-identified data will be available in the Mendeley database.
Trial registration number NCT05895643.
- Patients
- Clinical Trial
- Ethanol
- Functional Magnetic Resonance Imaging
- Substance misuse
- Randomised Controlled Trial
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- Patients
- Clinical Trial
- Ethanol
- Functional Magnetic Resonance Imaging
- Substance misuse
- Randomised Controlled Trial
STRENGTHS AND LIMITATIONS OF THIS STUDY
The trial is a double-blinded, randomised, placebo-controlled clinical trial, designed to evaluate the effects of the glucagon-like peptide-1 receptor agonist semaglutide on alcohol consumption in patients with alcohol use disorder and comorbid obesity.
The biological mechanisms will be investigated with multimodality brain MRI (magnetic resonance spectroscopy, functional MRI and diffusion tensor imaging).
Experienced nurses trained in cognitive behavioural addiction therapy will perform all therapy sessions, strengthening the standardised protocol for all included patients.
The alcohol biomarker phosphatidylethanol will be measured in plasma to support the self-reported alcohol consumption data.
It is a limitation that no semaglutide placebo pens from the semaglutide manufacturer (Novo Nordisk A/S) are obtained, and consequently, patients must visit the research facility once a week to receive the assigned treatment in a blinded fashion.
Introduction
Background
Alcohol use disorder (AUD) is a relapsing brain disorder characterised by loss of control over alcohol intake, a negative emotional state when not consuming alcohol and compulsive alcohol behaviour, leading to relapse.1 Up to 50% of patients with AUD experience alcohol withdrawal symptoms such as tremors, anxiety and nausea; some require medical detoxification treatment.2 Globally, alcohol use is a huge burden, and an estimated 280 million people suffer from AUD globally, making it one of the leading causes of preventable deaths.3 Despite this, the treatment gap is wide compared with other mental health disorders4—a Danish trial has shown an all-cause cumulative 10-year mortality rate of 29% after a first-time hospital contact due to alcohol.5 The AUD-associated high mortality rate is related to medical illnesses and traumas.1 5 6 From this perspective, AUD has severe consequences for individuals, relatives6 and society due to higher demand for health care5 and socioeconomic costs.2 3 This is why alcohol is considered to be the most harmful drug of addiction when both the harm to the user and to others are considered.7
Treatment of alcohol use disorder
AUD is a heterogeneous disorder, and several behavioural and psychological treatments are available.2 Cognitive behavioural therapy (CBT) is among the treatments with the highest empirical support.8 According to clinical guidelines, patients diagnosed with mild AUD are recommended psychological intervention, but an add-on of pharmacological treatment is recommended for patients with moderate to severe AUD.9 10 Only three pharmacological treatments (disulfiram, acamprosate and naltrexone) are approved by both the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA). A fourth medication, nalmefene, is only approved by the EMA.11 The pharmacological treatments for AUD play an essential role. However, there remains a need for more efficacious medication, and given the diverse biological processes contributing to AUD, the identification of novel molecular targets is needed to provide a broader catalogue of treatment options.
Glucagon-like peptide 1
In the search for new treatments for AUD and other addictive drugs, one focus has been on the incretin hormone glucagon-like peptide 1 (GLP-1).12–15 GLP-1 is an endogenous 30-amino acid peptide hormone produced by cleavage of the prohormone proglucagon. GLP-1 is produced in the L-cells in the small intestine and is released in response to food intake. With a half-life of only 3–5 min, it is rapidly inactivated by the enzyme dipeptidyl peptidase 4.16 GLP-1 regulates overall glycaemic control by stimulating insulin secretion and inhibiting glucagon secretion. GLP-1 also decreases gastric emptying and reduces appetite and food intake via stimulation of GLP-1 receptors in the hypothalamus, hindbrain and brain areas involved in reward.17 18 Importantly, GLP-1 is also produced in the nucleus tractus solitarius (NTS) of the brainstem. It is released as a neurotransmitter in the ventral tegmental area (VTA) and nucleus accumbens (NAc)19 and GLP-1 receptors are expressed in brain regions involved in addiction.20–23
Glucagon-like peptide 1 (GLP-1) receptor agonists and obesity
GLP-1-based therapy was introduced to the market in 2006, and since then, several GLP-1 receptor agonists (RA)s have been approved for treating type 2 diabetes and two for obesity. In 2014, the first GLP-1RA, liraglutide, was approved for treating obesity (body mass index (BMI≥30 kg/m2)).24 The investigational drug semaglutide was approved for treating type 2 diabetes in 2017. In 2021, semaglutide was also approved by the FDA and EMA for obesity treatment and was shown to be superior to the previous GLP-1RA liraglutide approved for weight loss.25 The reported anti-obesity effects of GLP-1RAs24 might be due to a change in food preference,26 satiety signal17 or the fact that individuals with obesity have deranged GLP-1 signalling.27 Overlapping dysfunctional brain circuits have also been observed in individuals with obesity or addiction,28 and a functional MRI (fMRI) study in obese versus lean individuals has reported normalisation of the brain response to a food paradigm in obese patients when treated with the GLP-1RA exenatide.29 Several preclinical studies report that several GLP-1RAs can cross the blood-brain barrier,30–32 and the investigational drug semaglutide is shown to reach the brainstem, septal nucleus and hypothalamus.30
Preclinical and clinical studies on the effects of GLP-1 receptor agonists on the consumption of alcohol and drugs of abuse
The potential alcohol-reducing effects of GLP-1RAs in humans were first described in a conference abstract in 2011, where a cross-sectional review conducted on patients from India with type 2 diabetes treated with liraglutide for 3 months reported a reduction in alcohol intake.33 However, those data have never been published in a journal format. Several first-generation GLP-1RAs, for example, exenatide, have shown promising results in preclinical addiction models in mice, rats and non-human primates,34–36 and the beneficial effects of the newer GLP-1RA semaglutide on alcohol intake are also described in recent preclinical trials.37–39 Preclinical addiction models in rodents have also shown that GLP-1RAs reduce the intake of nicotine, amphetamine, cocaine and heroin.15 40–42 As of now, only one randomised, placebo-controlled, double-blinded clinical trial investigating the effects of a GLP-1RA (exenatide) in patients with AUD has been published.43 This trial showed no differences in the reduction in heavy drinking days between the treatment groups. However, in an exploratory analysis of subgroups with and without obesity, alcohol intake was significantly reduced in the exenatide-treated group in the obese AUD patients but not in those without obesity.43 In addition, a subgroup of the total patient population had fMRI performed while engaging in an alcohol cue reactivity task, and the exenatide group had reduced ventral and dorsal striatum activation, compared with the placebo group.43 These fMRI data may imply that patients with AUD treated with exenatide experienced reduced incentive salience; that is, reduced sensitivity to alcohol-associated visual cues.44 Alcohol cue reactivity was also significantly reduced in the septal area, a brain area playing a pivotal role in reward45 and with a high expression of GLP-1 receptors.23 Further supporting a potential role for GLP-1RAs in AUD treatment, a recent cohort study suggested that the use of GLP-1 analogues prescribed for treating diabetes and obesity was associated with a lower incidence of alcohol‐related events.46 In addition, a case series of six individuals treated with semaglutide for weight reduction has reported a decrease in AUD symptomatology,47 a social media study reported reduced craving and desire to consume alcohol while in treatment with a GLP-1RA or GLP-1/gastric inhibitory polypeptide agonist48 and lastly secondary analysis from a study investigating the effects of the GLP-1RA dulaglutide on nicotine dependence reports a 29% alcohol reduction after 12 weeks of treatment.49
GLP-1, dopamine and GABA
Dopamine is considered to play a central role in the rewarding and addictive properties of drugs of addiction and alcohol,50 and GLP-1-containing neurons that project from the NTS to the VTA and the NAc have been identified.19 Several preclinical studies have tried to elucidate how GLP-1 systems modulate dopamine signalling. Gamma-aminobutyric acid (GABA) is an inhibitory neurotransmitter that modulates dopaminergic homeostasis,51–53 and preclinical and clinical studies have documented the involvement of the GABAergic transmitter system in AUD.51 54 55 Imaging studies using magnetic resonance spectroscopy (MRS) have shown that acute ethanol administration in healthy individuals reduces cortical brain levels of GABA52 and that GABA-A receptors are downregulated after prolonged ethanol withdrawal in patients with AUD compared with healthy individuals.51 Preclinical studies have shown that GLP-1 receptors are localised on cortical glutamatergic and GABAergic nerve terminals in rats56 and mice.53 Moreover, the GLP-1RA exenatide is reported to increase cortical and hippocampal GABA release, and the GLP-1 receptor antagonist exendin-3 can abolish these effects.53 In preclinical experiments, the GLP-1RA semaglutide is reported to modulate central GABA transmission.37
In addition to the reward circuitry, we also hypothesise that GLP-1 modulation of the habenula, which expresses high levels of GLP-1Rs57 and has been shown to modulate nicotine reward and intake,58 will also play a role in modulating alcohol consumption. Indeed, a recent study showed that exenatide delivered into the habenula reduced alcohol intake in rodents,59 and the habenula has been implicated in the aversive effects of alcohol.60
Hypotheses
Treatment with semaglutide compared with placebo will:
Decrease alcohol consumption in patients with AUD and comorbid obesity.
Reduce fMRI blood-oxygen-level-dependent response in reward processing regions during an alcohol cue task, including the septal area, and increase brain GABA levels measured by MRS in the anterior cingulate cortex.
Improve white matter tract integrity measured with a diffusion tensor imaging (DTI) scan.
Improve functional connectivity measured with resting state fMRI, including reducing connectivity between the habenula and the ventromedial prefrontal cortex.
Methods and analysis
Trial design
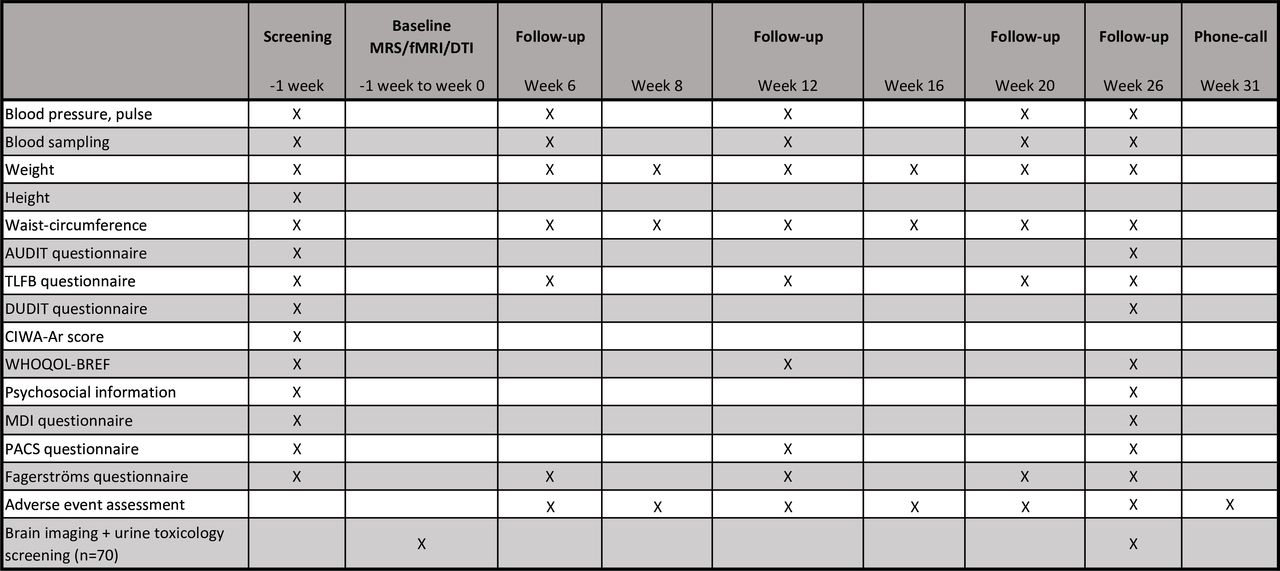
The present trial is a 26-week, randomised, double-blinded, placebo-controlled, single-site clinical trial designed to evaluate the effects of semaglutide versus placebo in 108 patients diagnosed with AUD and comorbid obesity (BMI ≥30 kg/m2). All patients will, in addition to the assigned treatment, receive 10 CBT sessions by experienced nurses trained in addiction cognitive therapy. A subgroup of the patients (n=70) will have brain imaging (MRS, resting state fMRI, fMRI alcohol cue task and DTI) performed at baseline and after 26 weeks of intervention. For safety evaluation, all included patients will be contacted by telephone 5 weeks after the last visit (figure 1).

AE, adverse events; AR, adverse reactions; DTI, diffusion tensor imaging; fMRI, functional MRI; MRS, magnetic resonance spectroscopy; SAE, serious adverse events; SAR, serious adverse reactions; sc., subcutaneous; TLFB, timeline follow back
Participants and screening
Patients will be recruited from general practitioners, hospital units or social nurses in the Capital Region of Copenhagen, Denmark, through our project webpage www.alkoholforskning.dk, and the patient recruiting webpage www.forskningnu.dk. All potential participants will be informed of their rights verbally and in writing by a medical doctor before signing the written consent form, and they must perform a breath alcohol test below 0.5‰. At the screening assessment, the patient will be asked about their alcohol intake in the previous 40 days, and this will be recorded via the validated timeline followback (TLFB) method.61 The patient will also be asked about their alcohol preference, previous treatment for AUD and general information about psychosocial factors, that is, education level, employment, marital status and the name plus contact information of a close relative. Height, weight, waist circumference, blood pressure and pulse will be registered. The following questionnaires will be applied: the Clinical Institute Withdrawal Assessment of Alcohol Scale-Revised (CIWA-Ar), Major Depression Inventory (MDI), Alcohol Use Disorder Identification Test (AUDIT), Drug Use Disorder Identification Test (DUDIT), Penn Alcohol Craving Scale (PACS), Fagerström Test for Nicotine Dependence (FTND) and WHO Quality of Life Score (WHOQOL-BREF). Daily medications and current somatic symptoms, that is, gastrointestinal symptoms, will be registered to obtain somatic baseline information. Safety blood samples will be analysed, and blood samples will be saved for an investigational biobank and a biobank for future research (independent consent form) (figure 2). The 2-hour assessment will take place at Mental Health Centre Copenhagen, Frederiksberg, Copenhagen University Hospital, Denmark.

{kind=link}
{kind=link}
The first injection of the assigned treatment is given at week 0. Cognitive behavioural therapy is provided in 10 sessions during the 26 weeks of inclusion. AUDIT, alcohol use disorders identification test; CIWA-AR, Clinical Institute Withdrawal Assessment for Alcohol-revised; DUDIT, Drug Use Disorders Identification Test; fMRI, functional MRI; MDI, major depressive inventory; PACS, Penn Alcohol Craving Scale; TLFB, timeline followback; WHOQOL-BREF, WHO Quality of Life Bref.
Intervention, blinding and safety
The pharmacological intervention with semaglutide or placebo will be combined with standardised cognitive behavioural therapy. Semaglutide is delivered from the pharmacy of the Capital Region of Copenhagen in a pre-filled FlexTouch pen-injector at doses of 0.25 mg, 0.5 mg, 1.0 mg, 1.7 mg and 2.4 mg. The placebo will be supplied as sterile pre-filled saline syringes (BD PosiFlush) and administered in the same way and volume as semaglutide. Needles are matched with the needle size of semaglutide. Both semaglutide and placebo will be administered once weekly subcutaneously by unblinded personnel not involved in other trial activities. The unblinded personnel will send individual text messages the day before the injection to remind the patients of their scheduled appointment. The personnel will also monitor trial adherence, which is defined as no more than three consecutive missed injections or six missed injections in total during the 26 weeks. All patients included will have a fictional prescription in their medical record, preventing other doctors from prescribing semaglutide. The initial dose of semaglutide is 0.25 mg once weekly, and the titration will follow the manufacturer’s recommendations until the maximal tolerable dose (2.4 mg) is achieved. To maintain the blinding of the patients, the preparations for the injection are performed in a separate locked room before the visit. The pens ready to inject are kept in a small individual opaque box for each patient, and the lid is only open when the patient is blindfolded by use of a sleeping mask. In order to avoid hearing the sound from the semaglutide pen when the injection is given, patients will wear headphones with music during the injection session. The unblinded personnel will perform the injection on the back of the upper arm. When the patient has left the research facility, the injection needle is discarded, and the pen is placed in the individual box until the next visit. This method has been used before, in our former alcohol GLP-1 study, without any unintentional unblinding.43 Patients, study personnel, other caregivers and persons performing the data analysis will remain blinded until all primary data have been analysed. In case of a serious adverse reaction requiring the knowledge of the assigned treatment, the randomisation code can be broken for that particular patient. Adverse events will be recorded once weekly and evaluated together with the safety blood samples by a blinded medical doctor and a team of specialists in psychiatry and endocrinology. If a patient is hospitalised, a project medical doctor and the trial sponsor are informed at the time of the admission, to evaluate potential serious adverse events (SAEs)/serious adverse reactions (SARs). Patients will also receive a phone number with 24-hour access to a blinded medical doctor. According to Danish law, no patients will have any economic incentives to stay in the trial.
Key inclusion criteria
Able to provide oral and written informed consent
Age 18–70 years
Diagnosed with alcohol dependence according to the criteria of the International Classification of Diseases 10th Revision (ICD-10) and AUD according to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5)
AUDIT score>15
BMI≥30 kg/m2
Heavy alcohol drinking here is defined as more than 6 days with daily alcohol consumption of four units (48 g alcohol) or more for women and five units (60 g alcohol) or more for men during a consecutive 30-day period within 40 days prior to the baseline evaluation, measured by the TLFB method. The 30-day period will be the 30 consecutive days with the largest alcohol intake (most heavy drinking days and the largest amount of total alcohol) out of the 40 days registered.
Key exclusion criteria
Severe mental illnesses, defined as having a diagnosis of paranoid psychosis, schizophrenia, bipolar disorder, or intellectual disability
A history of delirium tremens or alcohol withdrawal seizures
No serious withdrawal symptoms at inclusion (a score higher than nine on the CIWA-Ar scale
Present or former neurological disease, including traumatic brain injury
Type 1 diabetes, or type 2 diabetes in poor glycaemic control (defined as haemoglobin A1c (HbA1c) ≥48 mmol/L or fasting plasma glucose above 7.0 mmol/L at inclusion)
Females of childbearing potential who are pregnant, breastfeeding or have the intention of becoming pregnant within the next 9 months (26 weeks plus 2 months after discontinuation of semaglutide), or are not using contraceptives (during the whole study period) considered as highly effective (combined (oestrogen and progestogen containing) hormonal contraception associated with inhibition of ovulation (oral, intravaginal, transdermal), progestogen-only hormonal contraception associated with inhibition of ovulation (oral, injectable, implantable) intrauterine device (IUD), intrauterine system (IUS), bilateral tubal occlusion, vasectomised partner, sexual abstinence)62
Pregnancy (serum human chorionic gonadotropin (hCG)>3 U/L at inclusion) Impaired hepatic function (liver transaminases>3 times the upper limit)
Impaired renal function (estimated glomerular filtration rate (eGFR)<50 mL/min and/or plasma creatinine>150 µmol/L)
Impaired pancreatic function (any history of acute or chronic pancreatitis and/or amylase>2 times the upper limit)
Former medullary thyroid carcinoma (MTC) or Multiple Endocrine Neoplasia syndrome type 2 (MEN 2) or family history with MTC/MEN2.
Cardiac problems defined as decompensated heart failure (NYHA class III or IV), unstable angina pectoris or myocardial infarction within the last 12 months
Uncontrolled hypertension (systolic blood pressure>180 mm Hg, diastolic blood pressure>110 mm Hg)
Concomitant pharmacotherapy against AUD, that is, disulfiram, naltrexone, acamprosate or nalmefene, since the first day of the 30 days in the TLFB-drinking period registered at inclusion
Receiving any investigational drug within the last 3 months
Use of weight-lowering pharmacotherapy within the preceding 3 months
Any other active substance use indicated as a DUDIT score>1 (except nicotine)
Hypersensitivity to the active substance or any of the excipients
Only for patients undergoing brain scans:
Contraindications for undergoing an MRI scan (magnetic implants, pacemaker, claustrophobia, etc)
Unable to speak and/or understand Danish
Any condition that the investigator feels would interfere with trial participation.
Withdrawal criteria
Patients can withdraw from the trial at any time without providing a reason. Patients who discontinue the intervention and have been included for at least 12 weeks will be encouraged to participate in a premature week 26 follow-up, including brain scans. Failure to comply with trial medication, that is, if the patient misses more than three consecutive injections or more than six injections in total, leads to exclusion.
Timeline followback (TLFB) method
At weeks 0, 6, 12, 20 and 26, the validated TLFB method61 will be completed for the last 40 days in close collaboration between the patient and the investigator. The period with the 30 consecutive days with the largest alcohol intake (most heavy drinking days and the largest amount of total alcohol intake) will be recorded (figure 2).
Blood analyses
At weeks 0, 6, 12, 20 and 26, various standardised safety blood samples will be collected to evaluate liver, kidney and pancreatic function. HbA1c will be measured at weeks 0, 12 and 26 to evaluate glycaemic status. At weeks 0 and 26, plasma and serum will be collected for proteomics and plasma semaglutide measurements. At weeks 0, 6, 12, 20 and 26, plasma phosphatidylethanol (PEth)—the biomarker with the best correlation to self-reported alcohol intake prior to sampling63 will be collected. Alanine aminotransferase (ALT) and aspartate transaminase blood samples will be collected at weeks 0, 12 and 26 for the calculation of the Fibrosis-4 score (FIB-4).64 Whole blood is collected at weeks 0 and 26 in our biobank for future research (figure 2).
Questionnaires
Several questionnaires will be administered at weeks 0, 6, 12, 20 and 26 to assess potential psychopathology, life quality, other drug use, smoking status and the impact of alcohol: quality of life (WHOQOL-BREF), MDI, alcohol craving (PACS) and rating scales related to consumption of alcohol (AUDIT), tobacco (FTND) and other drugs (DUDIT) (figure 2).
Cognitive behavioural therapy
The patients will receive ten sessions of standardised CBT65 by a blinded experienced nurse trained in CBT. During the sessions, psychoeducation, motivation, drinking goals, self-control, behavioural analysis of drinking risk situations, general problem-solving (cravings and feelings), relapse prevention and lifetime alcohol use (ALCO-Life) will be assessed.
Brain imaging
To evaluate the neuroanatomical underpinnings and possible central mechanisms of action of semaglutide, non-invasive brain imaging will be performed at weeks 0 and 26 in a subgroup of 70 patients included in the trial. A Siemens 3 Tesla scanner placed at the Neurobiology Research Unit, Copenhagen University Hospital, Rigshospitalet, Denmark, will be used. The session will last 90 min in total and include fMRI to assess resting state functional connectivity,66 fMRI to assess reactivity to alcohol cues,67 MRS to measure GABA levels in the anterior cingulate cortex68 and DTI to evaluate structural connectivity.69
Sample size calculation and randomisation
The power calculation is based on the primary endpoint, that is, the change in heavy drinking days. No other studies have reported the effects of semaglutide in patients with AUD. The sample size calculation is based on the alcohol trial by Bogenschutz et al,70 exploring psilocybin-assisted therapy as a novel treatment of AUD. They showed a reduction in the total number of heavy drinking days of 47% in the intervention group and 25% in the placebo group. With a power of 90%, an alpha of 5% and an estimated SD of 26.44, we will need 64 patients, with 32 patients in each treatment group. However, large dropout rates are often observed in AUD intervention trials, with attrition rates between 10% and 35%.71 We anticipate a 40% dropout rate, making the total sample size 108, with 54 patients in each treatment group. The randomisation will be performed by unblinded personnel, using the randomisation module in REDCap72 stratified by age (two levels), gender (two levels) and baseline heavy drinking days (four levels). The block sizes will be randomised evenly between 2 and 4. The allocation sequence is generated at the webpage www.sealedenvelope.com and uploaded into REDCap by unblinded personnel. An unblinded personnel will inform the unblinded study personnel about the allocated treatment.
Endpoints
The primary endpoint is a change in alcohol consumption, defined as the change in the percentage of heavy drinking days during 30 consecutive days after 26 weeks of treatment adjusted for baseline, that is, percentage points (pp). A heavy drinking day is here defined as an intake of 60/48 g or more of alcohol in 1 day (men/women, respectively), recorded with the TLFB method. The secondary endpoints are change in heavy drinking days adjusted for maximum semaglutide dose, change in heavy drinking days adjusted for weight loss, changes in total alcohol consumption (gram ethanol/last 30 days), change in number of days without alcohol consumption (last consecutive 30 days), change in drinks per day (last consecutive 30 days), time to relapse defined as the time to first alcohol intake, time to first heavy drinking day, change in WHO alcohol risk level (last consecutive 30 days), change in alcohol craving (PACS score), change in AUDIT score, change in DUDIT score, change in liver fibrosis (FIB-4 score), change in life quality (WHOQOL-BREF), change in smoking habits (FTND) and change in blood parameters, gamma-glutamyl transferase (GGT), ALT, mean corpuscular volume (MCV) and PEth. In addition, changes in body weight (kilograms), blood pressure, pulse, waist circumference and change in glycaemic parameters (HbA1c). In addition to these clinical outcomes, brain GABA levels and fMRI alcohol cue reactivity will be explored in a subgroup of patients (n=70) at baseline and after 26 weeks of treatment.
Data management
All patients will receive a trial number when included in the trial. The patients will be informed orally and in writing that all data will be entered continuously and stored in a secure database with restricted access (REDCap).73 All data will be analysed anonymously in a statistical software programme according to national laws. The electronic database REDCap, source data, source documents, protocol and amendments, drug accountability forms, correspondence, patient identification list, informed consent forms and other essential good clinical practice (GCP) documents will be retained at the trial site for ten years after the trial.
Data analysis plan
All statistical analyses will be performed with treatment groups still blinded, and a detailed statistical analysis plan will be uploaded to ClinicalTrials.gov before starting the data analysis process. The statistical analyses will be performed using ‘RStudio’ software.74 Continuous outcomes, eg, primary endpoint, all secondary outcomes, sensitivity and subgroup analyses, will be analysed with a linear model adjusted for baseline until the last observational endpoint, eg, the week 26 follow-up. Continuous data will be summarised with the number of observations, mean, SD, minimum, median and maximum. Categorical data will be summarised with numbers and percentages. The level of statistical significance will be with an alpha set at 0.05, two-sided testing. There will be no adjustment for multiplicity, and no additional adjustments for covariates, except baseline values, will be performed. All analyses will be performed after the intention-to-treat principle; for example, all included patients who are randomised and receive at least one dose of the trial compound (semaglutide or placebo) will be included. There will be no replacement for patients dropping out. Missing data will be imputed using the multiple imputations method,75 and sensitivity analysis will be performed to evaluate the imputed results with complete cases, eg, a per-protocol analysis, and for the outcomes of heavy drinking days, total alcohol intake and days without alcohol consumption, we assume the dropout is caused by a relapse in alcohol intake, and therefore, we will impute the values of the missing week-26 data with a) the baseline values and b) half the baseline values. Withdrawal data will be presented in a Kaplan-Meier survival curve. Data from patients screened but not randomised to treatment will be illustrated in the CONSORT flowchart. Safety data will be collected during the 26 weeks of inclusion and 5 weeks after trial termination; that is, contacted by telephone by the investigators. All collected safety data will be summarised in a table with incident cases.
Ethics and dissemination
The trial is approved by the Ethics Committee of the Capital Region of Denmark/The Danish National Board of Health (EU CT NUMBER: 2023-503371-25-00) and the Danish Data Protection Agency (P-2023–187). The trial can be identified by ID NCT05895643 at ClinicalTrials.gov and UTN-number: U1111-1286-6919. The trial will be conducted in accordance with the Declaration of Helsinki II and with national laws and regulations for clinical research. The sponsor is responsible for informing the ethics committees and regulatory authorities of any SAEs or amendments to the protocol as per national requirements. The GCP unit of Copenhagen University, for example, the Danish Data Monitoring Committee, will monitor the trial. The clinical trial will comply with the protocol, according to ICH E6: Good Clinical Practice: Consolidated guideline, CHMP/ICH/135/95, and national guidelines, including Regulation (EU) No 536/2014. The hospital insurance covers all patients included. One or more manuscripts will be prepared for publication in high-impact international scientific journals. All results, that is, positive, negative and inconclusive results, will be published. The published international guidelines for authorship (International Committee of Medical Journal Editors, 1997) will be adhered to for both Danish and foreign collaborators. Results will be presented as posters or oral presentations at national and international congresses. All de-identified data and the protocol will be available in the Mendeley database after the results are published.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors According to the International Committee of Medical Journal Editors (ICMJE) criteria, all authors qualify for authorship. MKK and AF-J made the first draft of the trial protocol. All authors have contributed to the trial design. MKK, AF-J and CE performed the statistical power calculations. MKK, AF-J, GM, HB, NV and GMK undertook the final magnetic resonance spectroscopy/functional MRI/diffusion tensor imaging session design. MKK wrote the first draft of the manuscript. UBK is the trial sponsor. TK and JNP conducted the therapy sessions. MKK, SKJ, TK and JNP performed the screening and follow-up assessments. LR and SKJ conducted the brain imaging sessions. All authors contributed with critical revision of the manuscript for intellectual content and have approved the final manuscript. AFJ is the guarantor.
Funding This work is financed by the Mental Health Services, Capital Region of Copenhagen, the Novo Nordisk Foundation, the Novavi Foundation, the Augustinus Foundation, and the Hartmann Foundation. The funding sources and the manufacturer of semaglutide once weekly (Wegovy®, Novo Nordisk A/S) had no influence on the trial design.
Competing interests AF-J has received an unrestricted grant from Novo Nordisk A/S to investigate the effects of semaglutide on metabolic disturbances in patients with schizophrenia treated with antipsychotics and serves on a clinical trial advisory panel for Novo Nordisk (no honorarium). GMK has received personal honoraria from H. Lundbeck, Sage Biogen and Sanos, and chair for SIAB in HBP (personal honorarium). TV has served on scientific advisory panels, been part of speaker's bureaus, served as a consultant to and received research support from AstraZeneca, Amgen, Eli Lilly, Boehringer Ingelheim, Mundipharma, Gilead, MSD/Merck, Novo Nordisk and SunPharmaceuticals. HB has received personal honoraria from a Washington University Seminar. JJH has served on scientific advisory panels and/or speaker for Novo Nordisk, Eli Lilly and Zealand Pharma. He has given lectures and received financial support for travel from Novo Nordisk, Novo Nordisk Pharma, Novo Nordisk Scandinavia AB and Mayo Clinic. He has served as a consultant for Alphasights, Eli Lilly, Shouti/Structure TherapeuticsX and Zealand Pharma. He is currently consulting for GV Management LLC. He is the cofounder and on the board of directors of Antag Therapeutics and Bainan Biotech and sits on the board of directors of Antag Therapeutics and Bainan Biotech, which is unpaid. He is supported by grants from Arla Foods, ERC Advanced Grants and the Novo Nordisk Foundation Center for Basic Metabolic Research Faculty of Health and Medical Sciences University of Copenhagen, Denmark. He serves as an investigator for Boehringer Ingelheim and Scohia. GFM is a consultant for Merck & Co., Sumitomo Dainippon Pharma Co. and UCB Pharma. All other authors have no competing interests.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.