Article Text

Abstract
Introduction Pain is common in patients receiving mechanical ventilation in the intensive care unit (ICU). Intravenous opioids are recommended as first-line therapy for pain management; however, opioids have adverse side effects. Based on low-quality evidence, low-dose ketamine is therefore recommended as an opioid adjunct to reduce opioid consumption. Esketamine is an alternative to ketamine with greater efficacy and fewer side effects. However, evidence on the use of esketamine in patients receiving mechanical ventilation is lacking. This study investigates the efficacy and safety of esketamine as an adjunct to sufentanil for analgesic therapy in non-surgical ICU patients under mechanical ventilation.
Methods and analysis This ongoing multicentre, single-blind, randomised controlled trial is being conducted at six ICUs in China. 132 non-surgical patients under mechanical ventilation will be randomly assigned to the standard care and S-ketamine groups at a 1:1 ratio. Patients in the standard care group received a minimal dose of sufentanil as the sole analgesic agent. Patients in the S-ketamine group received a minimal dose of sufentanil in addition to an esketamine infusion at a fixed rate of 0.2 mg/kg/hour for analgesia. The primary outcome is mean hourly sufentanil consumption during the treatment period.
Ethics and dissemination This study was approved by the Ethics Committee of Chongqing University Cancer Hospital (CZLS2022067-A). Participants are required to provide informed consent. The results of this trial will be reported in peer-reviewed journals and presented at conferences.
Trial registration number ChiCTR2200058933.
- Adult intensive & critical care
- PAIN MANAGEMENT
- Randomized Controlled Trial
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
This is a multicentre, randomised controlled trial to evaluate the efficacy and safety of esketamine as an adjunct to sufentanil for analgesic therapy in non-surgical intensive care unit (ICU) patients under mechanical ventilation.
The study population is limited to non-surgical patients, which helps reduce bias caused by postoperative pain.
The study is single-blind only.
The study population is limited to non-surgical ICU patients under mechanical ventilation who do not require deep sedation or neuromuscular blockers, which may limit the generalisability of the results to other ICU patients.
The effect of esketamine on chronic pain and postintensive care syndrome will not be assessed.
Introduction
Pain is inevitable in mechanically ventilated patients in the intensive care unit (ICU) and is associated with poor outcomes.1–3 Intravenous opioids are recommended as first-line therapy for pain management.4 However, opioids have troublesome side effects, such as unexpected sedation, delirium, respiratory depression and ileus.5 A prior prospective cohort study demonstrated that adverse drug reactions in the surgical ICUs were mainly caused by opioids, which increased the length of ICU stay by 53.2%.6 Furthermore, the use of opioids in non-surgical ICU patients is associated with persistent opioid use.7 8 There is a growing concern that new persistent opioid use may be contributing to the ‘opioid crisis’.9–11
Current guidelines suggest that non-opioids should be used as adjuncts in ICU analgesia to reduce opioid consumption.5 However, commonly used non-opioids, such as acetaminophen and non-steroidal anti-inflammatory drugs (NSAIDs), may aggravate pre-existing organ dysfunction in critically ill patients.12–15 Acetaminophen-induced hypotension is common in critically ill patients, and acetaminophen hepatotoxicity is the leading cause of acute liver failure.13 16 NSAIDs have a weak opioid-sparing effect but may increase the risk of acute kidney injury (AKI) and gastrointestinal bleeding.17–19 Nefopam has significant opioid-sparing effects; however, there is a risk of increased heart rate and mild decrease in mean arterial pressure in critically ill patients.18 20 Moreover, nefopam is not available in many places outside Europe.
Ketamine is an N-methyl-D-aspartate receptor inhibitor with a short half-life and minimal adverse effects on the respiratory and circulatory systems and is used for anaesthesia and analgesia. Anaesthetic doses of ketamine can cause side effects such as hallucinations and cognitive impairment, while low-dose ketamine has good analgesic effects with fewer side effects than anaesthetic doses of ketamine.21 Based on the limited evidence obtained in surgical patients, the guidelines recommend the adjuvant use of low-dose ketamine to reduce opioid consumption.5 However, the analgesic effect of ketamine in ICU patients under mechanical ventilation remains controversial, especially in non-surgical patients.22 23 Esketamine (S-ketamine) is a right-handed enantiomer of ketamine with three times the potency of R-ketamine and twice that of racemic ketamine.24 Esketamine may reduce opioid consumption in patients outside the ICU and has fewer side effects than ketamine.25–28 To our knowledge, there are no studies demonstrating the efficacy and safety of esketamine for analgesia in non-surgical ICU patients under mechanical ventilation. This study was designed to assess whether esketamine can reduce opioid consumption and the associated clinical outcomes in non-surgical ICU patients under mechanical ventilation.
Methods and analysis
Design
This is a multicentre, single-blind, randomised controlled trial. The study design followed the Standard Protocol Items: Recommendations for Interventional Trials guidelines (online supplemental table S1). A flow chart of the study is shown in figure 1. This is version 1.2 of the protocol from 18 May 2022.
Supplemental material

Study flow chart. ICU, intensive care unit.
Setting
This ongoing study is being conducted at the ICUs of tertiary hospitals, including Chongqing University Cancer Hospital, Jinling Hospital, Fujian Provincial Hospital, Longyan First Hospital Affiliated to Fujian Medical University, Linyi City People Hospital, and Jiangsu Province Hospital of Integrated Chinese and Western Medicine.
Eligibility criteria, recruitment and informed consent
Inclusion criteria
Age between 18 and 70 years.
Non-surgical patients, defined as not undergoing surgery classified as grade 2 or above within 1 week, according to the ‘Management Measures for Surgical Grading in Medical Institutions’ established by the Chinese Ministry of Health.
Patients were intubated and mechanically ventilated within 12 hours and expected to require ventilation for longer than 48 hours.
Exclusion criteria
Pregnant or breastfeeding.
Medical condition preventing the assessment of Richmond Agitation-Sedation Scale (RASS) and Critical-Care Pain Observation Tool (CPOT).
Contraindications to esketamine hydrochloride.
Contraindications to sufentanil, propofol, midazolam or their excipients.
Requires deep sedation (RASS ≤−4) or continuous infusion of neuromuscular blocker or both.
Suspected or proven acute primary brain injury (traumatic brain injury, cerebral infarction, intracranial haemorrhage, spinal cord injury, hypoxic ischaemic encephalopathy, hydrocephalus or cerebral oedema).
Ejection fraction <30%, cardiogenic shock and acute myocardial infarction.
Endogenous creatinine clearance rate <30 mL/min.
End-stage liver disease (Child-Pugh grade C).
Requires surgery or tracheotomy within 48 hours.
Ketamine or esketamine hydrochloride required for status epilepticus or other diseases.
History of drug or alcohol abuse or both.
Palliative care or expected to die within 48 hours.
History of dementia or mental illness, or requires psychotropic medication.
Refusal to sign the informed consent form.
Participating in clinical trials of other drugs, or having participated in other clinical trials within 30 days.
Does not require opioids for analgesia, as determined by the clinician.
Recruitment and informed consent
The objectives, potential risks and benefits of this trial will be presented to the patients or their surrogate decision-makers. Randomisation and study intervention will begin after obtaining written informed consent. If written informed consent cannot be obtained within 12 hours of intubation, randomisation and timely intervention may start when verbal consent is obtained from the patients or their surrogate decision-makers, and written consent will be obtained later. The translated patient consent form is attached as online supplemental file 1.
Randomisation, allocation and concealment
Permuted block randomisation stratified by study site was used in this trial. Block lengths ranging from four to eight were used. Random allocation was performed using Interactive Web Response Systems (IWRS). Eligible patients were randomly allocated at a ratio of 1:1 to the standard care group and the S-ketamine group. Before being randomised, the choice of analgesic and sedative drugs and dose titration were determined by the patient’s treatment physician. Since the randomisation is done through the IWRS, the investigators, the treatment teams and the patients will not know the allocation until randomisation is completed. In this way, the allocation concealment is ensured.
Blinding
The grouping of interventions will be kept strictly confidential to patients until the results of the study are revealed. Several specialised personnel (blind assessors) will be established at each study site for CPOT and RASS assessments, and the study groupings will remain confidential to them. The treatment team will titrate the analgesics and sedatives according to the blind assessors’ assessments, in accordance with the study protocol and their experience. In addition, the study grouping will maintain the confidentiality of the outcome assessors and the trial statisticians who analysed the data.
Interventions
Administration and dosage adjustment of analgesics
Standard care group
In the standard care group, a minimal dose of sufentanil is used as the sole analgesic for pain management. The recommended sufentanil loading dose is 0.1–0.5 µg/kg, with an initial dose of 0.3 µg/kg/hour and a range of 0.15–0.7 µg/kg/hour, which can be adjusted at the discretion of the patient’s treating physician. Sufentanil was titrated to the minimum dose required to maintain the analgesic goal. The analgesic goal is to maintain CPOT ≤2. An intravenous bolus of sufentanil is allowed when there is a procedure or treatment ordered by a physician. CPOT will be reassessed every 2–4 hours, and the dose of sufentanil will be adjusted based on the CPOT assessment. Other analgesic measures (such as massage, music and relaxation techniques) for the standard care group will follow international guidelines and determined by the treating physician.5
S-ketamine group
In the S-ketamine group, esketamine hydrochloride (Hengrui Pharmaceutical) was infused at a rate of 0.2 mg/kg/hour in addition to the minimal dose of sufentanil for pain management. Sufentanil would be administered in the same manner as that in the standard treatment group, and esketamine hydrochloride will be administered within 1 hour of randomisation. CPOT will be reassessed every 15–30 min after the administration of esketamine hydrochloride. If CPOT is ≤2, the dose of sufentanil will be reduced by 10%; if CPOT is >2, the dose of sufentanil will be increased by 10%; this process will be repeated and the dose of sufentanil will be titrated to the minimum dose that can maintain a CPOT of ≤2. The esketamine hydrochloride dose will remain unchanged throughout the study period. Unless the patient remains oversedated when the administration of sufentanil and sedatives has stopped, esketamine hydrochloride will be reduced in a gradient of 0.05 mg/kg/hour until the sedation goal is achieved. The analgesic goals and other analgesic measures in the S-ketamine group will be the same as those in the standard care group. The analgesic dosing algorithm for the S-ketamine group is shown in figure 2.

Analgesics dosing algorithm of the S-ketamine group. CPOT, critical-care pain observation tool.
Duration of the intervention
For the standard care group, the intervention will stop when the following events occur (whichever occurs first): (1) 72 hours after randomisation; (2) analgesics not required due to extubation or other medical reasons, as determined by the treatment team; (3) the patient dies; (4) the patient requires surgery or tracheotomy; (5) the patient requires deep sedation or neuromuscular blockers; (6) treatment goals shift to palliative care; (7) severe adverse event occurred (see definition of severe adverse event in the Adverse events section); (8) the patient or family members withdraw informed consent; and (9) unable to accurately assess CPOT and RASS scores due to changes in disease status. For the S-ketamine group, in addition to the criteria for the standard care group, the intervention will also be discontinued if the criteria for discontinuation of esketamine hydrochloride are met. The criteria for discontinuation of esketamine hydrochloride are shown in online supplemental table S2.
Management of sedation, delirium, sleep disturbance and immobility
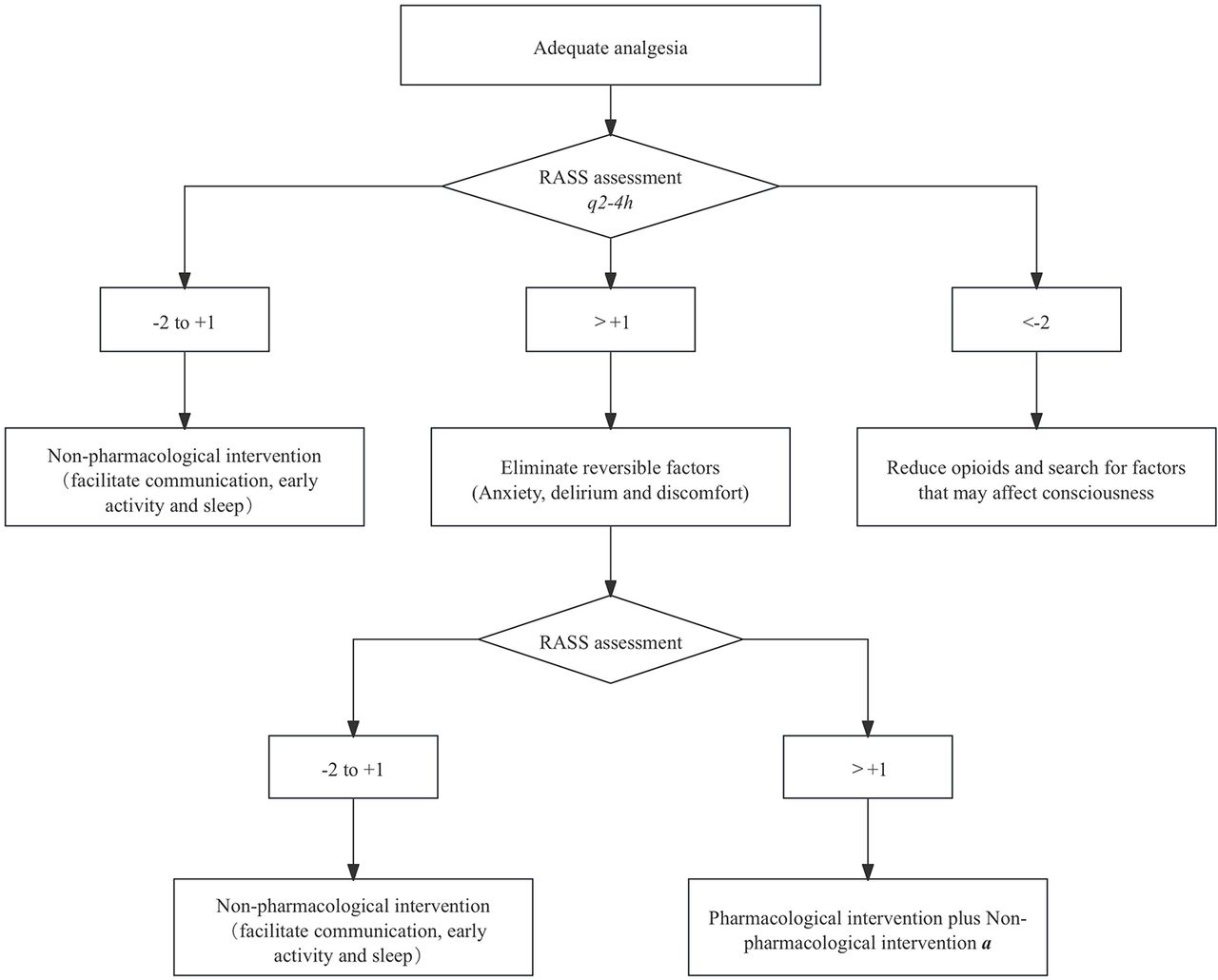
Management of sedation will follow the guideline recommendations and will be the same in both groups. The target RASS for both groups ranges from −2 to 1. Propofol is the sedative of choice, with midazolam as an alternative. Based on the guideline recommendations and the widely accepted early Comfort using Analgesia, minimal Sedatives and maximal Humanne care (eCASH) principle, a relevant sedation algorithm was established (figure 3). Management of delirium, immobilisation and sleep disturbance will follow the recommended guidelines.

{kind=link}
{kind=link}
{kind=link}
Sedation algorithm. aIn this study, propofol (loading dose 5 μg/kg/min, maintenance dose 5–50 μg/kg/min) was used as the preferred sedative, and midazolam (loading dose 0.01–0.05 mg/kg, maintenance dose 0.02–0.1 mg/kg/hour) was used as the alternative sedative. Other sedatives include cycloprofen (loading dose 0.1 mg/kg, maintenance dose 0.3-0.8 mg/kg/hour) and lorazepam (loading dose 0.02–0.04 mg/kg, maintenance dose 0.01–0.1 mg/kg/hour). Dexmedetomidine (no loading dose, 0.2–0.7 μg/kg/hour) was not recommended as the main sedative.RASS, Richmond Agitation-Sedation Scale (RASS) is a tool for assessing the depth of sedation in patients, which contains 10 levels, each corresponding to a different state of consciousness.
Mechanical ventilation and weaning
Mechanical ventilation will be implemented according to practical guidelines.29 Mechanical ventilation in patients with Acute Respiratory Distress Syndrome (ARDS) should follow the lung-protective ventilation strategy, including the use of lower tidal volumes (4–8 mL/kg predicted body weight) and lower inspiratory pressures (plateau pressure <30 cm H2O).30
Weaning from mechanical ventilation will follow the practical guidelines for mechanical ventilation.29 The specific processes include weaning screening, spontaneous breathing test (SBT), airway patency assessment and airway protection ability assessment. Patients who pass the SBT with good airway patency and protection will be weaned off and extubated.
Other therapies
It is not recommended to use analgesics other than sufentanil and esketamine hydrochloride in either group. Acetaminophen and NSAIDs can be used as antipyretics; however, their use should be recorded in detail. Nutritional therapy in both groups needs to follow the recommendations of relevant guidelines and the ‘Nutrition Support Process for Critically Ill Patients’.31 32 Treatment of the primary disease and comorbidities in both groups follows the corresponding guidelines and is determined by the medical team. Other symptomatic and supportive treatments determined by the patient’s medical team may also be provided.
Follow-up
All subjects will be followed for 28 days after randomisation or until the patient’s death, whichever occurs first. The indicators involved in the study are evaluated and recorded according to the study schedule (online supplemental table S3).
Data collection and management
A web-based database has been established for data collection, and the principal investigator and coinvestigator at each research site have access to the database. Data entered and modified by the investigators at each research site are based on the original data. The accuracy and compliance of the data will be audited by the principal investigator and coordinating centre. Once errors or omissions are found, specific personnel are asked to clarify the data and make corrections. The principal investigator will hold a training session for all coinvestigators involved in data collection before the commencement of the study to avoid inconsistencies in data collection. Range editing and value checking have been incorporated into the database to reduce data entry errors.
Outcomes measures
Primary outcome
Mean hourly sufentanil consumption during the treatment period, which is defined as the time from randomisation to the end of the intervention. The study intervention will be stopped according to the prespecified criteria outlined in the the duration of the intervention section.
Secondary outcomes
Mean hourly consumption of sedatives during the treatment period.
CPOT and RASS assessments every 4 hours during the treatment period.
Mean hourly consumption of sufentanil on the fifth day after randomisation.
Proportion of requiring frequent suctioning during the treatment period.
Proportion of uncontrolled agitation during the treatment period.
Sequential Organ Failure Assessment (SOFA) score on the first 7 days after randomisation (in ICU).
Acute Physiology and Chronic Health Evaluation II (APACHE-II) score on the seventh day after randomisation (in ICU).
Liver function, renal function and myocardial enzyme on the first 3 days and the seventh day after randomisation (in ICU).
Acute Gastrointestinal Injury (AGI) score, enteral nutrition tolerance score, gastric residual volume and intra-abdominal pressure on the first 7 days after randomisation (in the ICU).
Nutrition compliance rate on the fourth and seventh day after randomisation (in ICU).
Incidence of ICU delirium, number of delirium days and proportion of psychotropic drugs used for delirium (a positive Confusion Assessment Method of the Intensive Care Unit is considered as delirium).
Ventilation-free day in 28 days.
Vasopressor-free day in 28 days.
Adverse events, severe adverse events and adverse events that may be related to the study drug.
Length of ICU stay in 28 days.
Length of hospital stay in 28 days.
28-day mortality after randomisation.
Adverse events
Investigators and treatment teams will closely monitor possible and unexpected adverse events as well as severe adverse events during the trial. Adverse events are defined as any untoward medical occurrence in a patient who received an investigational intervention. A serious adverse event is defined as any serious medical event that causes death, life-threatening conditions, prolonged hospital stay, persistent disability or dysfunction, or other unpredictable serious medical events. The causal relationship between the adverse events and the study drug will be classified as certain, probable, possible, unlikely or uncertain. Any adverse events will be treated appropriately and recorded, and severe adverse events will be reported by the principal investigators. We have not set up a study-specific date monitoring committee for this trial. Alternatively, we are required to submit adverse events without assignment information to the institutional review board once a year.
Sample size calculation
Previous studies have shown that consumption of morphine in surgical ICU patients after major abdominal surgery was reduced by 25% in 48 hours (80±37 mg vs 58±35 mg) when ketamine was administered with an initial dose of 0.5 mg/kg followed by a perfusion of 2 µg/kg/min during the first 24 hours and 1 µg/kg/min in the following 24 hours.33 According to the single-center data from Chongqing University Cancer Hospital, the mean hourly consumption of sufentanil in mechanically ventilated patients may be reduced by about 26% (0.23±0.10 µg/kg/hour vs 0.17±0.09 µg/kg/hour) when a dose of 0.2 mg/kg/hour of esketamine hydrochloride adjunct to sufentanil is applied. We conservatively anticipate that the mean hourly consumption of sufentanil will decrease by 20% when a dose of 0.2 mg/kg/hour of esketamine hydrochloride adjunct to sufentanil is applied (μ1=0.94, μ2=0.75, σ=0.35). With a power of 90% and an α error of 0.05 (two-sided), a sample size of 120 subjects is needed, calculated using the PASS V.11.0 software: 60 in the standard group and 60 in the S-ketamine group. Considering the possibility of dropouts, a sample size of 132 study participants was planned (10% inflation), including 66 in the standard care group and 66 in the S-ketamine group.
Statistical analysis
The primary comparative analysis will be based on the intention-to-treat population, and secondary supportive analyses will be done on the Per-protocol population. The safety analysis will be performed on the safety population. Missing data will be handled by multiple imputations to evaluate the robustness of the primary endpoint analyses. The normality distribution of the variables was tested using the Shapiro-Wilk test. All numerical continuous variables will be presented by mean±SD or median±IQR according to whether they obey the normal distribution. Counting and categorical variables will be presented as proportions, frequencies or percentages. Normally distributed continuous variables will be statistically analysed using Student’s t-test, and non-normally distributed continuous variables will be statistically analysed using the Wilcoxon rank-sum test. Counts and categorical variables will be statistically analysed using the χ2 test or Fisher’s precision probability test. For the primary outcome, a generalised linear mixed model (GLMM) will be used to compare group differences. In the GLMM model, the mean hourly sufentanil consumption during the treatment period will be treated as the response variable following a Gaussian distribution and the esketamine intervention as the fixed effect and site as the random effect, and the identity link function will be used. Additionally, some prespecified variables were preplanned as covariates, including age, gender, body mass index and baseline APACHE II scores. From this model, the difference of the mean hourly sufentanil consumption during the treatment period and its two-sided 95% CI for the group comparison will be estimated. For the secondary outcomes, the GLMM models were also used, and the identity-Gaussian models will be used for continuous endpoints and log-binomial models for categorical endpoints. If the above log-binomial regression model does not converge, the Mantel-Haenszel method will be used to calculate the relative risk (RR) and its 95% CI stratifying by site. If the above continuous endpoints do not fulfil the normal distribution assumption of the models, data conversions (including log, reciprocal and square root transformations) will be performed. P values will be reported with two decimal points and all tests will be two-sided. P values with a level of significance of <0.05 will be considered statistically significant. No interim analysis was planned in our study.
Patient and public involvement
None.
Study status
The trial was registered on 20 April 2022, and the first patient was randomised on 23 June 2022. The planned end date for the study was originally 30 November 2023. However, due to various factors, the recruitment process was slower than expected. At the time of writing, 55 patients have been randomised and enrolment is ongoing.
Ethics and dissemination
This study was approved by the Ethics Committee of Chongqing University Cancer Hospital. The ethical approval document ID is CZLS2022067-A. The research sites obtained ethical approval from local ethics committees. Participants are required to provide informed consent. The results of this trial will be reported in peer-reviewed journals and presented at conferences.
Discussion
Although the guidelines recommend the use of multimodal analgesia to reduce the adverse effects of opioids, only approximately one-third of mechanically ventilated ICU patients are administered non-opioids for pain management.5 20 This partly relates to the adverse effects of currently used non-opioids and the lack of solid evidence; therefore, it is necessary to expand the analgesic arsenal and to provide stronger evidence. Esketamine has the potential to reduce opioid consumption. Several randomised trials that evaluated the analgesic effects of esketamine are ongoing outside the ICU.34–36 Song et al37 conducted a single-arm clinical study on esketamine in combination with remimazolam tosilate for analgesic sedation in mechanically ventilated ICU patients. To our knowledge, this is the first parallel randomised trial to evaluate the efficacy and safety of esketamine as an opioid adjuvant for analgesia in critically ill patients under mechanical ventilation.
Previous studies have revealed that high-dose ketamine causes anaesthesia and is associated with side effects such as hallucinations and delirium, whereas low-dose ketamine delivers promising analgesic effects and is less likely to cause side effects.38 Ketamine was considered as low dose at an infusion rate of 0.1–0.5 mg/kg/hour.23 33 39 40 Esketamine is theoretically twice as potent as racemic ketamine.25 Based on the abovementioned data, the fixed infusion rate of 0.2 mg/kg/hour of esketamine hydrochloride was adopted in this study, which is similar to the dose selected by Song et al.37
Per the guidelines, carefully titrated analgesic dosing is important to balance the benefits and potential risks of opioid exposure.5 In this study, the sufentanil dose was minimised to achieve the analgesic goal. Therefore, as in other studies, the mean hourly consumption of sufentanil was chosen as the primary outcome because it illustrates the analgesic effects of esketamine and reflects the potential benefits of reducing opioid consumption.23 33 41 Since different analgesic drugs and analgesic measures may affect analgesic and sedative effects in addition to patient outcomes, analgesic effects, organ function and other outcomes were included as secondary outcomes.42 43 Opioid agonists (particularly μ-opioid receptor agonists) are associated with gastrointestinal motility dysfunction.44 It has been demonstrated that ketamine-based anaesthesia reduces gastrointestinal inhibition compared with fentanyl-based anaesthesia, and food intake improves when analgesic sedation was switched to ketamine.45 46 Thus, our study assessed whether esketamine had a similar effect. Finally, the adverse effects of ketamine in mechanically ventilated patients in the ICU are of great concern and are controversial, and the common adverse effects of ketamine (such as delirium and increased secretions) were also included as secondary outcomes.23 47
The time window for randomisation and initiation of the intervention in this study was narrow enough to be in line with clinical practice and guidelines.23 27 48 49 Given the short intervention period of the study, delayed administration of the study drug may lead to false-negative results; therefore, esketamine was required to be administered within 1 hour after randomisation.
The following measures were taken to control for bias in this study:
Randomisation and allocation concealment were done.
The study population was limited to non-surgical patients to avoid the impact of postoperative pain.
Patients with factors that may affect drug response and analgesic evaluation were excluded.
The treatment team and the scoring assessors were independent. The assessors were unaware of the study groupings and the treatment team adjusted the dosage based on the assessors’ results. All patients followed the same analgesic approach (using the minimum dose of sufentanil to achieve the analgesic goal).
A dose adjustment algorithm was developed for the S-ketamine group to avoid bias caused by differences in analgesic modulation at different study sites.
The study protocol was discussed and training was conducted to ensure that the protocol was fully understood and strictly implemented by the researchers at each participating site.
This study had some limitations. First, this is not a double-blind study. Although efforts have been made to control bias, the single-blind design of the study could not completely avoid bias. Second, the study population was limited to non-surgical ICU patients under mechanical ventilation who did not require deep sedation or neuromuscular blockers, which may limit the generalisability of the results to other ICU patients. Third, the effect of esketamine on chronic pain and postintensive care syndrome was not observed in this study. Nevertheless, recent investigations have indicated that ketamine might be associated with chronic pain.50
In conclusion, the results of this trial may reveal whether low-dose esketamine can reduce opioid usage in non-surgical patients under mechanical ventilation and whether it is associated with clinical improvements.
Supplemental material
Ethics statements
Patient consent for publication
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors All authors (YL, DH, HN, DZ, TZ, SL, XL, QL, RL, ZJ and LK) were involved in the study design. YL, DH, HN, DZ, TZ, SL, QL and RL are responsible for carrying out recruitment, managing the treatment of the patients and collecting data. YL, LK and ZJ drafted the protocol and wrote the manuscript. All authors (YL, DH, HN, DZ, TZ, SL, XL, QL, RL, ZJ and LK) have read and edited the manuscript and approved the submission of the final manuscript. YL is responsible for the overall content as guarantor.
Funding This study was supported by the Chongqing Joint Medical Scientific Research Project of Science and Health (grant number 2020FYYX091) and Chongqing Science and Technology Committee (grant number cstc2017shmsA130057). The study protocol was peer-reviewed by the funding bodies. The funding bodies had no role in the design of the study and will have no role in the data collection, analysis, interpretation or reporting of the results.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.