Article Text

Abstract
Introduction Oral pre-exposure prophylaxis (PrEP) is a highly effective HIV prevention method; however, uptake and persistence have been low among southern African women. A dual prevention pill (DPP) that combines PrEP with oral contraception (OC) may increase PrEP use and better meet women’s sexual and reproductive health needs. We will gauge the DPP’s acceptability in two cross-over clinical trials.
Methods and analysis PC952 (Zimbabwe) and PC953 (South Africa) will compare acceptability, adherence and preference for an over-encapsulated DPP versus PrEP and OCs taken separately. HIV-negative, non-pregnant cisgender females in Johannesburg, South Africa (n=96, 16–40 years) and Harare, Zimbabwe (n=30, 16–24 years) will be randomised 1:1 to the order of regimens—DPP or two separate tablets—each used for three 28-day cycles, followed by a 6-month choice period in South Africa. Monthly clinic visits include HIV and pregnancy testing; safety assessments and risk reduction and adherence counselling. We will assess adherence (monthly) based on tenofovir diphosphate drug levels in dried blood spots and by self-report. We will evaluate acceptability (monthly) and preference (end of cross-over) via computer-assisted self-interviewing and in-depth interviews with a subset of participants. Data collection started in September 2022 and ended in January 2024.
Ethics and dissemination PC952 was approved by the Ministry of Health and Child Care, Medical Research Council, Research Council and Medicines Control Authority of Zimbabwe; the Chitungwiza City Health Ethics Committee; and the Joint Research Ethics Committee for the University of Zimbabwe Faculty of Medicine and Health Sciences and Parirenyatwa Group of Hospitals. PC953 was approved by the South African Health Products Regulatory Authority and the University of the Witwatersrand’s Human Research Ethics Committee. The Population Council IRB approved both studies. We will disseminate results in open-access journals, clinical trials registries, and at local and international meetings and conferences.
Trial registration numbers NCT04778514, NCT04778527.
- HIV & AIDS
- clinical trial
- decision making
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THESE STUDIES
The cross-over design enables a direct comparison between the intervention (DPP) and standard of care or control (oral pre-exposure prophylaxis (PrEP) and combined oral contraceptives (COCs) separately) regimens that enables a smaller sample size with women serving as their own controls.
Participants will be followed monthly, which will enable frequent assessments of PrEP drug levels in blood, however, it will not be feasible to measure contraceptive drug levels in blood due to the short half-life of COCs.
The two pilot studies enable early assessments of the DPP in two different contexts: South Africa (low COC use/high HIV incidence and prevalence) and Zimbabwe (high COC use/moderate HIV incidence and prevalence) and among adolescent girls and young women and older women.
The key limitation of the studies is the use of an overencapsulated DPP as a proxy for the ultimate coformulated tablet, which may not accurately capture acceptability, preference and adherence for the ultimate DPP, which will be a smaller tablet versus a capsule.
Another limitation is the different study designs and sample sizes for the two studies, primarily related to their respective funding mechanisms, which may limit the ability to directly compare results from the two countries.
Introduction
Despite substantial advances in HIV treatment and prevention over the last decade, women and girls in eastern and southern Africa continue to be disproportionally affected by HIV/AIDS, accounting for 63% of all new HIV infections in the region in 2021.1 In 2021, new HIV infections in South Africa were almost double among women aged 15 and over than among men of the same age (130 000 vs 70 000, respectively).2 Similarly, in Zimbabwe, nearly twice as many women 15 and older acquired HIV in 2021 compared with their male peers.1
Oral pre-exposure prophylaxis (PrEP) is more than 90% effective in reducing HIV transmission.3 However, many oral PrEP trials and demonstration projects in sub-Saharan Africa have been plagued by low adherence, particularly among adolescent girls and young women (AGYW).4 5 Stigma and fear of intimate partner violence or relationship dissolution are often cited as reasons for non-use of PrEP.6–9 Novel strategies to bolster uptake and adherence are needed to increase PrEP use among women and girls at high risk of HIV.
Many women—and AGYW in particular—are more worried about unintended pregnancy than HIV.10 11 Furthermore, there is a growing body of evidence indicating that many women may be more likely to use an HIV prevention method that also prevents pregnancy.12–19 Condoms are currently the only multipurpose prevention technologies (MPTs) that prevent both HIV and unintended pregnancy.20 Male condoms, however, are not under a woman’s control; female condoms have had limited uptake due to access and acceptability issues21 22; and many women risk gender-based violence by merely suggesting condom use.23 Several novel MPTs are in the pipeline,24 including a dual prevention pill (DPP) containing the ingredients in oral PrEP and oral contraception. The first DPP being developed is based on the commonly used 28-day combined oral contraceptive (COC) regimen (150 µg levonorgestrel (LNG), 30 µg ethinyl estradiol (EE)) and a generic equivalent of Truvada (300 mg tenofovir disoproxil fumarate (TDF), 200 mg of emtricitabine (FTC)).25 26 The DPP is likely to be the fastest route to an approved MPT as it contains two registered products that are safe and effective for their respective indications27–30 with no evidence of drug–drug interactions.31–34 In South Africa, 25.6% of women report ever having used COCs and 10.5% currently use them.35 36 In Zimbabwe, COCs are the most common family planning (FP) method among those using a modern method.37 Data on oral contraceptive use among adolescents in sub-Saharan Africa are limited; however, an analysis from 33 sub-Saharan African countries indicated that COC use ranged from approximately 15%–20% among 15–24 year-olds.38 Truvada is approved as PrEP in more than 20 countries globally, including South Africa and Zimbabwe, and is recommended by WHO and CDC ((US) Centers for Disease Control and Prevention) for women at risk of HIV using COCs.29 39
We hypothesise that the DPP could greatly increase PrEP adherence, while also meeting women’s unmet FP needs. Our goal is to generate data to inform DPP introduction through two clinical cross-over studies comparing acceptability of, adherence to and preference for the DPP versus two separate tablets. To that end, and in parallel with the development of the coformulated DPP, we have overencapsulated PrEP (Truvada) and a COC (Zinnia F (150 LNG/30 EE)), into a single capsule (figure 1) for our studies to provide an early indication of DPP acceptability.

Overencapsulated dual prevention pill.The hand shown is of one of the coauthors and is not a patient.
Methods and analysis
Trial design
(The Standard Protocol Items for Randomised Trials reporting guidelines is used.) Population Council Protocols 952 and 953 are randomised, controlled, open-label, cross-over studies.
Participant and public involvement
Formative research (December 2020–June 2021) with service providers and potential end users in South Africa and Zimbabwe informed the clinical trial design, materials and recruitment methods. Established community and/or youth advisory groups at each site reviewed the protocols and provided input into the consent forms, behavioural questionnaires and translations. The study teams have also benefited from participation in the DPP consortium, a collaborative group of researchers, donors and civil society advocates established to inform and accelerate DPP development and introduction.40
Study settings
Hillbrow (Johannesburg), South Africa
PC953 is being conducted at the Wits RHI (Wits Reproductive Health and HIV Institute) Research Centre, a large research clinic situated in Hillbrow, Johannesburg. As of 2020, Johannesburg had 756 751 people living with HIV with an overall HIV prevalence of 13%. HIV prevalence was highest among females across all age groups: 28.3% for 25–49 years, 15% for women 50 and above and 9% for 15–24 years.41 Wits RHI conducts research on HIV, sexual and reproductive health (SRH) and vaccine-preventable diseases.
Chitungwiza (Harare), Zimbabwe
PC952 is being implemented by the University of Zimbabwe Clinical Trials Research Centre (UZ-CTRC) at the Zengeza Clinical Research Site (CRS) in Chitungwiza, Zimbabwe’s second-largest city approximately 30 km south of Harare, with a generalised epidemic and HIV prevalence of 3.8% among women aged 15–19 years and 6.4% among women aged 20–24 years.42 The Zengeza CRS is located within a Chitungwiza City Health Department Municipal Clinic and conducts research on female-controlled HIV/STI (sexually transmitted infection) prevention strategies, including microbicides, oral and injectable PrEP and cervical barriers, and integrated HIV prevention strategies.
Study populations
We are recruiting participants (n=96, 16–40 years, South Africa; n=30, 16–24 years, Zimbabwe) from FP, PrEP and SRH clinics, and the general population. Although the DPP may ultimately appeal to individuals using other contraceptives or with an unmet FP need, we are enrolling participants who are already using COCs and are accustomed to daily pill-taking and the associated side effects of COCs (though they will not be used to the large DPP capsule). In addition, a recent study in Cape Town found that only 52% of 15–19 years (n=50/96) randomly assigned to use COCs (vs intravaginal rings or injectables) reported being fully adherent over an 8-week period,43 highlighting the importance of enrolling participants who have already been using COCs for at least 3 months.
Inclusion/exclusion criteria
Eligible participants are healthy, HIV-negative, non-pregnant, sexually active, cisgender females at moderate to high risk of HIV infection. Specific eligibility criteria (table 1) for the two protocols are similar, with several differences based on routine national protocols for PrEP provision and age range. Screening by nurses/clinicians includes medical history, physical examination and clinical laboratory tests. HIV risk is assessed by clinicians using local PrEP guidelines and by participants who are offered access to tools, such as BWise,44 to help them make that determination. Key exclusion criteria and prohibited medications relate to contraindications for COCs or PrEP use. We are also excluding those unable to swallow a large vitamin pill similar in size to the overencapsulated DPP (figure 1).
Inclusion and exclusion criteria for South Africa (SA) and Zimbabwe (Zim)
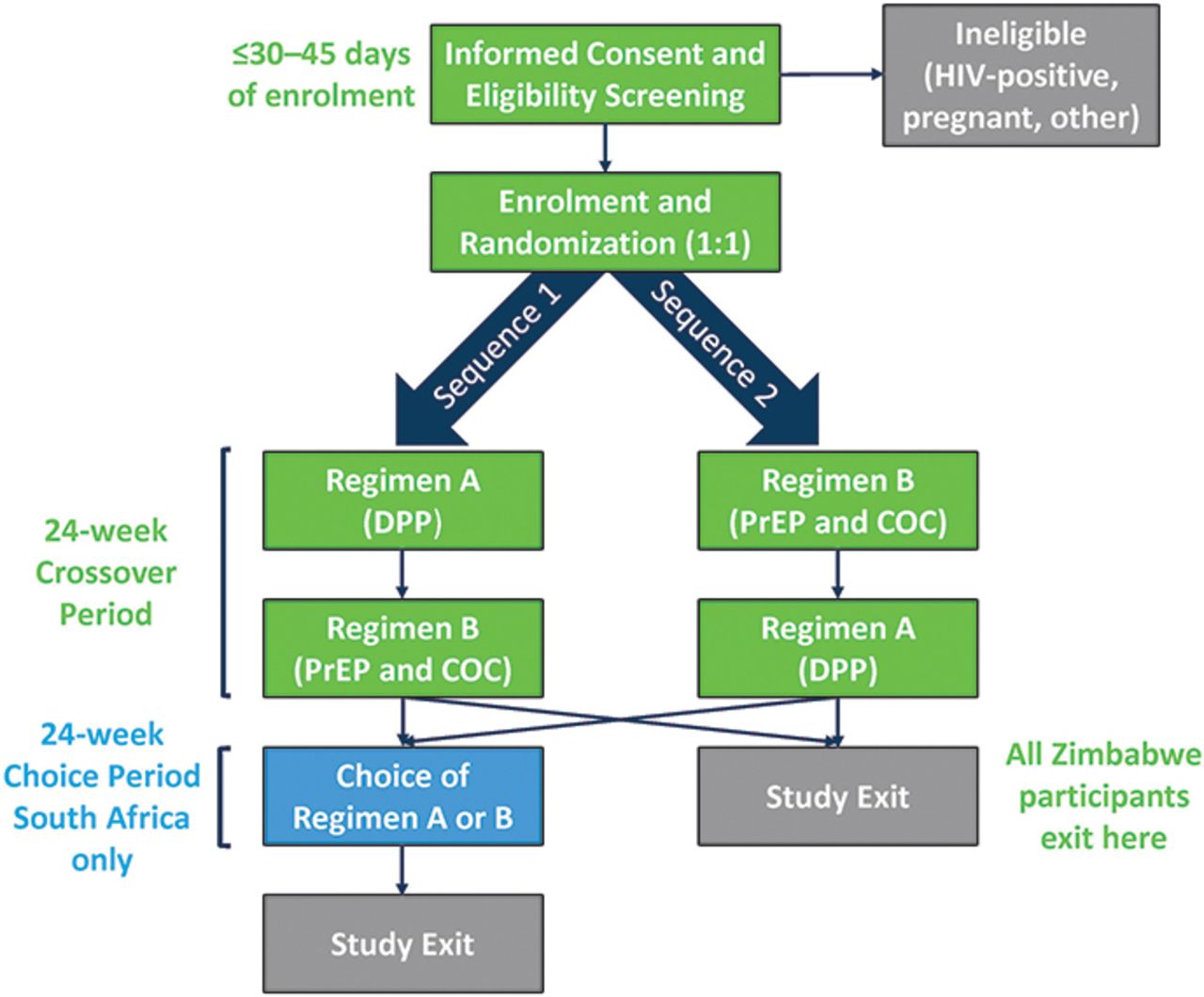
Study schema
At enrolment, participants are randomised to the sequence of study regimens (figure 2): sequence 1=single DPP capsule once daily for three 28-day cycles (regimen A), followed by two separate tablets (oral PrEP and COC) once daily for three 28-day cycles (regimen B); sequence 2=regimen B followed by regimen A. In South Africa, after the 6-month cross-over period, participants may choose regimen A, B or neither for up to six additional 28-day cycles. Study product regimens are described in detail in table 2.

{kind=link}
{kind=link}
Study schema. COC, combined oral contraceptive; DPP, dual prevention pill; PrEP, pre-exposure prophylaxis.
Study products
All participants take PrEP (FTC/TDF 200/300 mg) once daily by mouth throughout the entire study and 21 days of active COCs, followed by 7 days of placebo tablets, regardless of regimen. The selection and timing of doses correspond to the labels for Truvada (Gilead Sciences; Foster City, California, USA) and Zinnia F COCs (Mylan Laboratories, Hyderabad, India). Zinnia F was selected because it is the same formulation as Control L, the COC purchased for public FP programmes in Zimbabwe. During the ‘placebo’ days of regimen A, participants take one capsule containing Truvada only, whereas the placebo days of regimen B consist of two separate tablets: Truvada and a placebo COC tablet. The DPP capsules were manufactured and packaged by PCI Pharma Services (Rockford, Illinois, USA) from the same batches of Truvada and Zinnia F as the separate pills procured from Gilead and Mylan, respectively.
Study objectives
Objectives and outcomes are similar in both trials (table 3).
Objectives and endpoints for PC952 (Zimbabwe) and PC953 (South Africa)
Study procedures
Informed consent
Before undergoing screening procedures, a counsellor/designee leads a discussion to review the informed consent form in detail with potential participants in their preferred language (English or isiZulu, South Africa; English or Shona, Zimbabwe). The same study staff member implements a comprehension assessment to check participants’ understanding of key study aspects, including the potential increased risk of HIV or unintended pregnancy if difficulty swallowing the large DPP capsule leads to more missed doses, before they both sign the consent form. For unemancipated minors (16–17 years), informed consent from the parent/legal guardian is obtained before assent from the minor. Key elements of the informed consent are reviewed on an ongoing basis and willingness to continue study participation is ascertained.
Eligibility screening
After consenting, potential participants are assigned a unique participant identification number (ID) and undergo screening procedures. All screening test results and, if enrolled, study information (data, specimens) are recorded with IDs and no other identifying information to preserve participant confidentiality. Locator information is collected at screening and reviewed at each study visit to ensure participants are contactable for retention purposes. The screening process typically takes more than one day because several lab tests are outsourced. At screening (visit 0), a nurse/clinician takes a complete medical history, including gynaecological and obstetric history. A clinical exam is performed to assess overall health (including complete blood count (CBC) in Zimbabwe). Urine is tested for pregnancy (human chorionic gonadotrophin (hCG)), and Neisseria gonorrhoeae/Chlamydia trachomatis (nucleic acid amplification test (NAAT)). Blood is tested for HIV, syphilis and hepatitis B virus (and Hepatitis C in Zimbabwe), and to measure creatinine clearance. Screening also includes direct observation of participants swallowing a large vitamin capsule, similar in size to the 000 DPP capsule (figure 1).
Enrolment and randomisation
Enrolment (visit 1) is scheduled when participants are starting their next COC pack (±5 days). At enrolment, participants are tested for HIV (rapid antigen blood test) and pregnancy (urine hCG) to confirm eligibility. Those eligible are enrolled and randomised (1:1) to the sequence of regimens, are given a supply of their first study product with detailed dosing instructions and take their first dose directly observed in the clinic. Participants are counselled on management of anticipated side effects and missed pills based on recommendations that incorporate differing guidelines for COCs and oral PrEP.45 Participants also receive counselling on HIV/STI risk reduction, contraception and protocol compliance—including the importance of attending clinic visits and taking the study products—at every visit.
Follow-up visits
Online supplemental table 1 contains the detailed schedule of visits and procedures. At all visits, blood is collected for dried blood spots (DBS) to assess tenofovir-diphosphate (TFV-DP) levels as a measure of PrEP adherence46 and for rapid HIV testing. Urine is collected for pregnancy testing. After the first three cycles, participants ‘cross-over’ to their second regimen at month 3/visit 4 and return unused study product from their first regimen. They then receive their first supply of the second regimen, with detailed instructions, and take their first dose directly observed in the clinic. Participants attend monthly follow-up visits during the second regimen (month 4/visit 5; month 5/visit 6). At month 6/visit 7, all participants exit in Zimbabwe. In South Africa, participants may choose to continue using either regimen for up to another 6 months during a ‘choice’ period, with similar monthly visits.
Supplemental material
Laboratory procedures
Laboratory assessments are listed in online supplemental table 1. Blood specimens for hepatitis, creatinine, HIV confirmation testing (and CBC in Zimbabwe) are processed off-site by BARC (South Africa) and UZ-CTRC (Zimbabwe). DBS specimens are analysed at the University of Cape Town by liquid chromatography-tandem mass spectrometry.46
Safety monitoring and adverse event reporting
Clinical assessments at each visit post-enrolment are done to monitor potential adverse events (AEs) and social harms. Individual participants who develop grade 1 or unrelated grade 2 AEs, based on the Division of AIDS Grading system,47 may continue using their assigned study product(s) per protocol, at the site PI’s/designee’s discretion. Individuals who develop a related grade 2 AE, or any grade 3 AE, regardless of relatedness, will be evaluated by the site PI/designee and medical monitor for possible discontinuation from the study. Grade 4 AEs, regardless of relatedness, will be evaluated by the site PI/designee and Population Council Medical Monitor and those participants will be discontinued from the study. No dose modifications will be undertaken nor are there any a priori stopping rules because both study products (PrEP and COCs) are marketed drugs.
Seroconversion or pregnancy
Participants who seroconvert are terminated from the study. At their closing visit, study staff will collect unused pills, conduct resistance and viral load testing, and link the participant to HIV/FP care per local guidelines. Similarly, participants who become pregnant will be terminated and referred to services for pregnant individuals, including PrEP provision, if desired. The sites will make every effort to follow-up on all pregnancy outcomes. The sites may continue counselling participants as they transition to services to preserve their confidentiality after discontinuation.
Creatinine
Creatinine levels are monitored according to PrEP guidelines in each country, approximately quarterly.48 49 Participants with abnormal creatinine levels may be put on a temporary product hold, pending the PI/designee’s decision, until a repeat test can be done. Participants who have two tests outside the normal range will be permanently discontinued to reduce their risk if the DPP or PrEP is contraindicated.
Data and safety monitoring
The Population Council monitor conducted site qualification and initiation visits at both sites before data collection began. Periodic monitoring visits ensure the protocol and good clinical practice are being followed. The monitor reviews source documents to confirm that the data recorded on case report forms (CRFs) is accurate, and reviews relevant documents to verify protocol compliance. A data safety and monitoring board (DSMB) was established, consisting of three experts with clinical expertise in HIV and contraception, epidemiology, biostatistics and clinical trials. The DSMB (charter available on request) will review data after all participants are enrolled (both countries), and after all participants complete the cross-over visit (South Africa). All serious AEs (SAEs) and AEs leading to discontinuation will be reported to the relevant IRBs/ethics committees, drug regulatory authorities, sponsor, DSMB and funders. Unanticipated AEs that are potentially related to the study product(s) will be reported as suspected unexpected serious adverse reactions to the manufacturers (Gilead or Mylan).
Clinical staff are trained to identify, probe for, manage and report AEs and social harms at every visit. Study clinicians review abnormal test results, liaise with local clinic doctors and have the authority to terminate participants based on clinical opinion. On completion of the study, participants are referred to local clinics for PrEP and COC services, if they want to continue the methods. Any breaches in confidentiality, study protocol or AEs attributable to this study will be reported to the relevant IRBs/ethics committees and regulatory authorities.
Data collection and management
Clinical CRFs
CRFs were developed by the Population Council and the trial sites to capture demographics, medical history, clinical exam results, laboratory test results, product supply/pill counts, AEs, randomisation and termination data. Data are collected and managed using REDCap (Research Electronic Data Capture) hosted at the Population Council.50 51 Data are entered into REDCap within 5 days of each participant’s visit. Queries are triggered during data entry or by the Population Council data manager during weekly data reviews.
Quantitative behavioural surveys
At each visit, participants complete a behavioural questionnaire via computer-assisted self-interview (CASI) in their choice of English or the local language (isiZulu in South Africa, Shona in Zimbabwe). CASI questionnaires take approximately 30 min to complete and include questions about product acceptability, adherence and overall trial experiences. Participants complete their interviews privately on tablet computers, with study staff nearby to address potential technical challenges.
Qualitative exit interviews
A subset of participants will take part in an in-depth interview (IDI) after exiting the study. In South Africa, we will interview up to 30 participants representing those exiting early (during the cross-over period), those exiting after the cross-over period, and those completing the choice period (half who chose the DPP and half who chose two separate tablets). In Zimbabwe, we will interview all willing participants. IDIs will be conducted by female research assistants using a semistructured guide to explore preference for the DPP or two separate tablets; reasons for continuation/discontinuation; influence of partners, family and support structures; side effects; provider interactions and other factors affecting DPP acceptability and adherence. IDIs will last 40–60 min and will be scheduled at the closing visit or on a separate date, depending on participant availability. IDIs will be conducted in the participant’s choice of language; will be audiorecorded and transcribed; and translated into English (if necessary) for analysis.
Statistical considerations
Sample size and power calculations
South Africa
The sample size calculation was based on comparing adherence between the two regimens. A sample size of 86 has 80% power to detect a difference between the proportion of women who are adherent to each regimen assuming 25% of women are adherent to PrEP alone, 40% are adherent to the DPP, with a correlation between regimens of 50% and no period effect. We estimated 25% of participants would be adherent to the 2-pill regimen based on findings from other PrEP studies among young women in sub-Saharan Africa.5 52 We increased the sample to 96 in case 10% of participants discontinued early while still having 86 participants complete the cross-over period.
Zimbabwe
The sample size was calculated based on detecting a difference in preference for the DPP versus two separate pills. A sample size of 30 has 94% power to detect a preference for one regimen over the other when the true preference for one regimen is at least 80% based on the exact binomial test (alpha=0.04). If only 27 AGYW complete the study (10% loss to follow-up), we have 84% power to detect a preference for one regimen over the other when the true preference for one regimen is at least 80%, based on the exact binomial test (alpha=0.02).
Randomisation
Randomisation schemes for each study were developed by the Population Council biostatistician using SAS/STAT V.9.4 (SAS Institute Inc., Cary, North Carolina) with a 1:1 allocation using permutated block sizes. In South Africa, randomisation is in blocks of 12, 6 participants per sequence in each of 8 blocks. In Zimbabwe, randomisation is in blocks of 10, 5 participants per sequence in each of three blocks. The randomisation schemes are embedded within the REDCap systems for each study. At enrolment, the clinician consults REDCap to assign the treatment sequence for each sequentially enrolled participant.
Data analysis
The ‘all participants’ population includes all enrolled participants, the ‘safety population’ includes all participants who have used at least one dose of either regimen, and the ‘per-protocol’ population includes all participants who complete both regimens. In general, descriptive statistics (frequencies, mean, standard deviation, range) will be used to summarise and characterise data collated on differences in participants assigned to each sequence. Point estimates and corresponding two-sided 95% confidence intervals will be presented for endpoints, where appropriate. Missing data will not be imputed.
Preference
Preference for the DPP will be measured as the proportion of women (per-protocol population) reporting at the end of the cross-over period that they prefer the DPP capsule versus two separate tablets (or vice versa) by testing whether this proportion is greater than 0.5 using a z-test statistic under the exact binomial test in Zimbabwe (n=30) and normal approximation of the binomial distribution in South Africa (n=96). However, if the number of women completing each sequence is unbalanced, the comparison will be done using a random effects mixed model adjusting for effects treatment sequence may have on preference. In South Africa, we will similarly analyse the proportion of women who choose the DPP versus two separate tablets for the choice period.
Adherence
Adherence (overall) will be measured (safety population) by self-report, pill count and TFV-DP levels in DBS measured at each visit and will be compared by regimen. Adherence in DBS will be assessed by comparing the proportion of women with TFV-DP levels consistently greater than the threshold known to provide efficacy, using McNemar’s test for paired proportions. Adherence by pill count and self-report will be measured as the proportion of the total number of doses taken versus the total number of expected doses. If all women do not complete the cross-over period, analyses will be conducted with those who completed all six visits (24 weeks). Adherence during the choice period (South Africa) will be analysed similarly.
Acceptability
Acceptability of using the DPP capsule versus two separate pills will be measured in the safety population using a quantitative acceptability questionnaire. The primary outcome of acceptability will be measured based on responses to questions in the following acceptability domains: use attributes, product attributes, side effects and effect on sexual activity. Acceptability scores will be summarised by regimen and time point and compared by regimen at each visit. Scores will be compared using a random effects mixed model to evaluate the effects of regimen and time point.
Safety
Safety data include findings from physical (and pelvic, when indicated) exams, laboratory tests and AEs. AEs will be coded in accordance with the Medical Dictionary for Regulatory Activities.53 A summary of AEs will be based on treatment-emergent AEs, which include all AEs occurring on or after the first dose. The number and percent of participants for each AE and SAE will be summarised by system organ class and preferred term, overall and by regimen.
Effects of sociocultural and demographic characteristics
Effects of sociocultural and demographic characteristics (eg, age, education, income, employment, relationship status, HIV risk perception, self-efficacy for HIV prevention) on preference, acceptability and adherence will be explored using random effects mixed models. Sociocultural and demographic data will be collected at screening and enrolment.
Qualitative data
Qualitative data will be analysed thematically by researchers at the study sites and the Population Council. We will apply inductive (data driven) and deductive (a priori) codes to the data using software, such as NVivo. Coded data will be synthesised to generate descriptions of behaviours, attitudes and beliefs about the acceptability of the DPP capsule, preference for the DPP or two separate pills, trial experiences and other emergent themes.54–56
Ethics and dissemination
Ethics
Both protocols and amendments, informed consent forms and recruitment materials were approved by the Institutional Review Board of the Population Council (New York, New York, USA). The South Africa protocol and amendments (PC 953, V.3.0, 8 June 2022), consent forms, recruitment materials (where relevant) and data collection instruments in English (and isiZulu where relevant) were reviewed and approved by the University of the Witwatersrand Human Research Ethics Committee and South African Health Products Regulatory Authority. The Zimbabwe protocol and amendments (PC952, V.3.0, 17 June 2022), consent forms, recruitment materials (where relevant) and data collection instruments in English (and Shona where relevant) were approved by the Medical Research Council of Zimbabwe, the Medicines Control Authority of Zimbabwe, the Joint Research Ethics Committee of the University of Zimbabwe, the Ministry of Health and Child Care of Zimbabwe, the Chitungwiza City Health Ethics Committee and the Research Council of Zimbabwe. Both studies are being conducted in accordance with the US Code of Federal Regulations, the International Conference for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Guideline for Good Clinical Practice E6 (R2), and local standard operating procedures at each site. Participants are compensated for each visit commensurate with the norms and standards in each country. Both trials are registered on ClinicalTrials.gov (NCT04778527 and NCT04778514). Screening began in August 2022 and data collection ended in January 2024.
Dissemination
The study teams provide periodic updates to their communities and Community Advisory Boards during trial implementation. On completion, results will be presented locally at each site during in-person/virtual meetings with study participants, community advisory boards and other local stakeholders; at national and international conferences; through the DPP consortium; and posted on PrEP Watch. Manuscripts will be submitted to peer-reviewed journals and will be made available via open-access whenever feasible. Data will be uploaded on the ClinicalTrials.gov site, the US Agency for International Development’s Data Development Library (South Africa only) and in-country registries, as applicable. Datasets and protocols will be available from the sponsor on request.
Ethics statements
Patient consent for publication
Acknowledgments
We would like to acknowledge George Creasy, who spearheaded work to develop a DPP; Bruce Variano for overseeing the development and testing of the over-encapsulated DPP; Dan Loeven for overseeing regulatory aspects of manufacturing the DPP; Susanna Grecky and Rebecca Brodsky for liaising with suppliers and manufacturers; Sherry Hutchinson for creating the educational materials for the trial and graphics appearing in the paper and Heather Sussman for study oversight as clinical trial monitor.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @SanyuktaMathur
Contributors BF wrote both study protocols and the final version of the manuscript; SM conceptualised and wrote the behavioural data collection aspects of the protocol and manuscript; MP wrote the data management and statistical analysis sections of the protocol and manuscript; NM, AD, CM, PM, SM, TP-P, KR, NN, SK, LS, BZ and LH participated in protocol development and contributed to the manuscript. ML wrote an earlier draft of the paper; IB oversees study implementation, assisted in writing and editing the paper and prepared the manuscript for publication. All authors reviewed and approved the final version of the manuscript.
Funding PC953 is made possible by the generous support of the American people through the US President’s Emergency Plan for AIDS Relief (PEPFAR) and US Agency for International Development (USAID) via cooperative agreement #No. AID-OAA-A-13-00088. PC952 research reported in this publication was supported by the National Institute of Mental Health of the National Institutes of Health under Award Number R34MH119982. The Children’s Investment Fund Foundation (CIFF) Award Number 1903-03681, 2106-06589 supported the manufacturing of the study product, as well as supplementing the US government funding for both studies. Gilead donated the Truvada tablets for both studies.
Disclaimer The contents are the responsibility of the authors and do not necessarily reflect the views of PEPFAR, USAID or the US Government. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily reflect the views of CIFF.
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.