Article Text

Abstract
Introduction Emicizumab prophylaxis is approved for people of all ages with haemophilia A (HA) including infants and children. Although previous studies have demonstrated the efficacy and tolerability of emicizumab in infants with HA, real-world data on emicizumab use in infants are limited. The Haemophilia A in Infancy and NewbOrns: multi-instituional prospective observational study to assess the efficacy anD safety of Emicizumab (HINODE) study aims to evaluate the coagulation potential and safety of emicizumab prophylaxis in infants with congenital HA from birth to <12 months of age.
Methods and analysis This is a multicentre, observational study conducted in Japan in infants with congenital HA aged <12 months who are receiving or are scheduled to receive prophylactic emicizumab at an approved dosing regimen: 1.5 mg/kg weekly, 3 mg/kg every 2 weeks or 6 mg/kg every 4 weeks. The target inclusion is 50 infants. The primary endpoint is to evaluate the relationship between global coagulation test parameters (using clot waveform analysis and thrombin generation assay) and plasma emicizumab concentrations in infants aged from 6 to <12 months. Secondary endpoints include evaluating coagulation profiles in infants aged <6 months and changes between the age of <6 and 6 to <12 months. Additionally, coagulation parameters will be evaluated with the in vitro addition of anti-idiotype antibodies against emicizumab or the addition of a factor VIII product in infants aged from 6 to <12 months. The study will also evaluate adverse events and bleeds.
Ethics and dissemination The study was approved by the MINS Clinical Trial Review Committee (no. 230214) and will be conducted in compliance with the Declaration of Helsinki, the Act on the Protection of Personal Information and the Guidance of Ethical Guidelines for Medical and Biological Research Involving Human Subjects. Written informed consent for participation in the study will be obtained from a legally acceptable representative. Results will be published in scientific/medical journals and presented at international congresses.
Trial registration number Japan Registry of Clinical Trials; jRCT1031230264.
- HAEMATOLOGY
- Bleeding disorders & coagulopathies
- PAEDIATRICS
- NEONATOLOGY
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
STRENGTHS AND LIMITATIONS OF THIS STUDY
The HINODE study is a prospective, multicentre study in Japan with a target inclusion of 50 infants <12 months of age with congenital haemophilia A (HA) on emicizumab prophylaxis.
Global and general coagulation tests will be used to assess the relationship between coagulation potential and plasma emicizumab concentrations in infants with HA aged <12 months.
Changes in coagulation parameters during treatment with emicizumab will also be evaluated between the age of <6 months and 6 to <12 months.
As an observational study, there may be a low accumulation of cases <6 months of age.
Obtaining a sufficient volume of blood sampling is not easy in infants and the frequency of venous punctures should be kept at a minimum.
Introduction
Congenital haemophilia A (HA) is a hereditary condition characterised by a deficiency of coagulation factor (F)VIII; this manifests as recurrent bleeding, most commonly into joints and muscles which results in progressive joint health deterioration over the lifetime of a patient.1 Some types of bleeds can also be life-threatening including gastrointestinal or visceral organ bleeding and intracranial haemorrhage (ICH).1 The latter is a particular concern for neonates with approximately 2.1% of newborns estimated to experience an ICH in the neonatal period.2 The relative risk of ICH in neonates with haemophilia is 11.2 times higher than in infants aged 1–12 months with haemophilia3 and the cumulative mortality associated with ICH in neonates with haemophilia is estimated to be 0.2 per 100 live births2 with notable morbidity including neurodevelopmental problems, common in survivors.4 After the neonatal period, ICH is also observed in infants and toddlers.5
Regular prophylaxis should be started at a young age as a lack of adequate bleed prevention puts infants with HA at higher risk of developing life-threatening ICH and long-term joint health issues.1 6 It has been reported that just 2–3 bleeds into the same joint can lead to irreversible joint damage, ultimately reducing long-term quality of life.7 Early prevention and pre-emptive/presymptomatic treatment of bleeding in infants have traditionally involved intravenous infusion of an FVIII replacement product. However, the intravenous mode of administration and the frequency of infusions often necessitating the use of a central venous access device makes prophylactic FVIII replacement challenging in infants.8 9 Additionally, inhibitors to FVIII develop in approximately 30% of previously untreated people with severe HA, rendering it ineffective for bleed control.10 11
Emicizumab is a bispecific antibody that mimics the function of activated FVIII(a) binding to FIXa and FX to promote haemostasis in people with HA.12 13 It is administered subcutaneously as a loading dose of 3 mg/kg weekly (QW) for 4 weeks followed by maintenance dosing of 1.5 mg/kg QW, 3 mg/kg every 2 weeks (Q2W) or 6 mg/kg every 4 weeks (Q4W).14 15 Emicizumab is approved for prophylaxis for people of all ages with HA including infants and children irrespective of the FVIII inhibitor status.14 15 However, there is limited data on early emicizumab prophylaxis in infants with HA and the optimal point at which prophylaxis should be started in infants has not been established.
Neonates have an immature coagulation system at birth which evolves with age; it has been reported that several coagulation factors including FIX and FX, the target molecules of emicizumab, are present at approximately half of the adult levels at birth in healthy full-term infants and increase to the normal range by 6 months to 1 year after birth.16 17 Although the plasma concentrations of these coagulation factors are low at an early age, emicizumab has been shown to improve in vitro coagulation potential in plasma from infants and toddlers with HA.18 Additionally, previous clinical trials have demonstrated a favourable safety profile and effective bleed control in paediatric participants aged ≤12 years receiving emicizumab but only a few infants were included in these studies.19 20 Recently published results from the ongoing HAVEN 7 clinical trial have demonstrated the efficacy and tolerability of emicizumab in 55 previously untreated or minimally treated infants from birth to 12 months of age.21 However, real-world data on emicizumab use in infants with HA are scarce.22–24
Following the findings of the HAVEN 7 trial, the Haemophilia A in Infancy and NewbOrns: multi-instituional prospective observational study to assess the efficacy anD safety of Emicizumab (HINODE) study was designed to provide further clinical evidence on coagulation capacity and support the real-world safety of early prophylactic emicizumab in children with HA from just after birth to <12 months old. As the coagulability of emicizumab is difficult to assess directly in vivo in addition to the thrombin generation assay (TGA)25 used in HAVEN 7, this study will use clot waveform analysis (CWA)26 as global coagulation tests in participants. Furthermore, coagulation tests will be performed after the in vitro addition of anti-emicizumab idiotype antibodies27 or the addition of an FVIII product. Here, we report details from the HINODE study protocol (V.2.0; 1 June 2023).
Methods and analysis
Objectives
This study will evaluate the coagulability of emicizumab in infants with congenital HA aged <12 months. In addition, changes in coagulability under emicizumab treatment between <6 months and 6 to <12 months of age as well as the safety of emicizumab up to 18 months of age will be evaluated.
Study design and clinical setting
The HINODE study is a prospective, multicentre, unblinded, non-randomised, single-arm, observational study conducted only in Japan. The study is observational in nature as it will evaluate infants with congenital HA aged <12 months who are already receiving or are scheduled to receive prophylactic treatment with emicizumab. There will be no additional therapeutic interventions and limited invasiveness for blood collection. Emicizumab will be provided by the Japanese universal health insurance system which allows citizens to access a wide range of medical services. Once patients are diagnosed with HA, public funds will fully cover the necessary medical expenses.
The target inclusion in the study is 50 infants with congenital HA who have entered or are anticipated to enter the maintenance phase of emicizumab treatment (defined as having received≥5 doses) at <12 months of age and from whom blood samples can be collected during the maintenance phase while the infant is <12 months of age. Emicizumab will be administered by subcutaneous injection at one of the three approved dosing regimens in Japan: 1.5 mg/kg QW, 3 mg/kg Q2W or 6 mg/kg Q4W;15 treatment will continue at the discretion of the treating physician.
After enrolment in the study, blood samples for coagulation tests will be collected at the following time points: two times at the age of <6 months (Visit 1) and 6 to <12 months (Visit 2), respectively, in infants who have entered the maintenance phase of emicizumab treatment at the age of <6 months; or only once at the age of 6 to <12 months (Visit 2) in infants who have entered the maintenance phase of emicizumab treatment between the age of 6 and <12 months. All participants will have a safety follow-up assessment (Visit 3) at the age of 18±3 months. A summary of the HINODE study design is shown in figure 1.

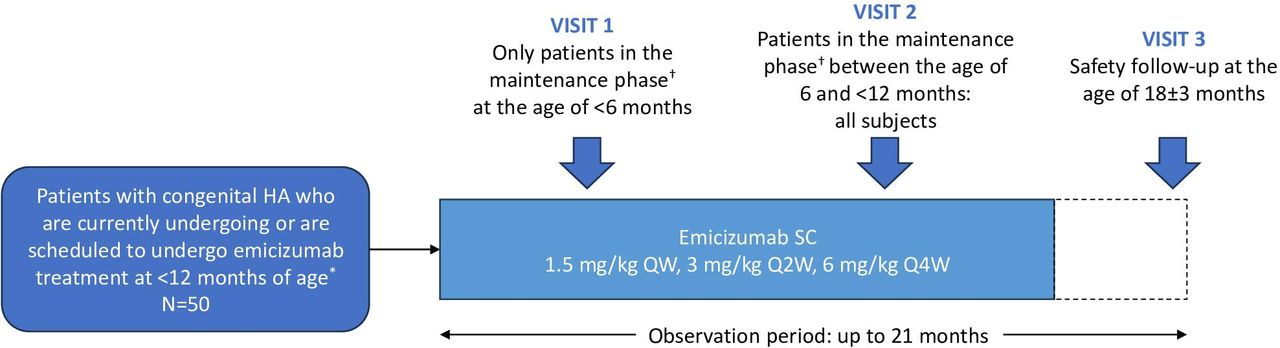{kind=link}
Study design. *Patients who have entered or are anticipated to enter the maintenance phase of emicizumab treatment and whose blood samples can be collected while they are <12 months of age. †The maintenance phase represents the fifth and subsequent doses of emicizumab. HA, haemophilia A; QW, weekly; Q2W, every 2 weeks; Q4W, every 4 weeks; SC, subcutaneous.
The study will continue until participants reach 18 months of age, until the date the investigator decides to terminate the study for that participant or until the participant’s legally acceptable representative requests to discontinue participation in the study. The final day of the study for each participant will be the last observation day, the day of the observation at the time of study discontinuation, the day on which consent is withdrawn or the day when the participant is lost to follow-up. The planned study period is from July 2023 to September 2027 with the planned duration of enrolment being 96 weeks from the day of the first enrolment and the planned duration of follow-up being up to 84 weeks from the day of the last enrolment.
Eligibility criteria
Inclusion criteria
For study entry, participants must have a diagnosis of congenital HA and must have emicizumab treatment selected as the most appropriate form of care. Additionally, they must be undergoing or must be scheduled to undergo treatment according to one of the dosage and administration regimens in the latest electronic package insert. Finally, participants must have entered or are anticipated to enter the maintenance phase of emicizumab treatment (≥5 doses) at <12 months of age with the possibility of collecting blood samples during the maintenance phase. Infants who receive FVIII at diagnosis before commencing emicizumab prophylaxis are eligible for the study. Written informed consent must be provided by a legally acceptable representative.
Exclusion criteria
Infants will be excluded from the study if they have an inherited or acquired bleeding disorder other than HA or if there is any reason that would render them unsuitable for study participation in the judgement of the investigator.
Endpoints
Primary endpoint
The primary endpoint is to evaluate the relationship of global coagulation test parameters (CWA and TGA) with plasma emicizumab concentrations in infants with congenital HA aged from 6 to <12 months, using CWA and TGA parameters from healthy individuals as reference values.
Secondary endpoints
Secondary endpoints include the evaluation of: (1) parameters of general coagulation tests including FVIII activity and of global coagulation tests with in vitro addition of anti-idiotype antibodies against emicizumab or addition of an FVIII product in infants with congenital HA aged from 6 to <12 months; (2) relationships of coagulation parameters (global and general coagulation tests including FVIII activity) with plasma emicizumab concentrations in infants with congenital HA aged <6 months; (3) changes in coagulation parameters between the age of <6 months and 6 to <12 months. If sufficient blood sample volume remains, general coagulation parameters in infants aged from 6 to <12 months and both general and global coagulation parameters in infants aged <6 months will also be evaluated under emicizumab neutralisation after in vitro addition of anti-idiotype antibodies against emicizumab and after addition of a FVIII product.
Additional secondary endpoints include evaluation of adverse events (AEs) (particularly development of FVIII inhibitors and abnormal laboratory values) and bleeds (particularly ICH) treated with a blood coagulation factor product.
Assessments
Blood samples will be collected for the evaluation of coagulation parameters at Visit 1 and Visit 2 or Visit 2 only depending on the age at which participants enter the maintenance phase of emicizumab treatment as mentioned in the study design section above. The frequency and volume of blood collection will follow the guidelines for sampling provided in the European Union guidelines on ethical considerations for clinical trials in the paediatric population.28 Where available, coagulation parameters will be compared between the two visits. Bleeds and medications used as well as AEs will be monitored throughout the study. The full schedule of assessments is shown in table 1.
Schedule of assessments
Blood samples collected at Visits 1 and/or 2 will be used for global coagulation tests (CWA and TGA) and general coagulation tests including FVIII activity, FIX activity, FX activity, activated partial thromboplastin time, prothrombin time, antithrombin, fibrinogen, D-dimer, fibrin degradation products and plasma emicizumab concentrations. Blood tests and analyses will be performed in central laboratories. Global coagulation activity will be assessed using adjusted maximum coagulation velocity (Ad|min1|) by CWA and peak thrombin by TGA. Plasma emicizumab concentrations will be measured using the r2 Diagnostics emicizumab calibrator and will be quantified as μg/mL of emicizumab.29 Additionally, in blood samples collected at age 6 to <12 months (Visit 2), CWA and TGA will be performed under emicizumab neutralisation by in vitro addition of anti-idiotype antibodies against emicizumab and after in vitro addition of a FVIII product and results will be compared against coagulation parameters obtained without additions. If sufficient blood sample volume remains, evaluation of coagulation parameters at age <6 months and other coagulation tests for age 6 to <12 months will also be performed under emicizumab neutralisation after in vitro addition of anti-idiotype antibodies against emicizumab and after addition of a FVIII product.
Data analysis
Sample size
The target sample size of 50 participants was not based on a statistical hypothesis and was selected in consideration of the following factors: the number of infants born with congenital HA in Japan, the number of emicizumab-treated infants with HA, the proportion of facilities providing emicizumab treatment participating in the study and the informed consent rate.
Data collection and monitoring
Data will be collected in electronic Case Report Forms (eCRFs) via electronic data capture (EDC). Study sites will be responsible for data entry into the Liscio/EDC system (Precursor Corporation, Japan). A subject ID number will be assigned to participants by the study sites to maintain confidentiality. Central monitoring will be performed based on the entered data collected in the eCRFs to confirm that the study is being conducted safely and per the protocol and that data are being accurately collected. On-site auditing will be considered if necessary to improve the scientific and ethical quality of the study.
Safety evaluations
Once emicizumab treatment begins, all AEs occurring up to the last observation day, the day on which safety follow-up is completed at the age of 18±3 months, the day of discontinuation, the day on which consent is withdrawn or the day on which the participant is lost to follow-up are subject to reporting and must be recorded in the eCRF regardless of the relationship to emicizumab. Investigators will seek information on AEs at each participant contact and update the information in the eCRF. All AEs whether reported by the participant’s legally acceptable representative or noted by study site personnel will be recorded in the participant’s medical records. The WHO toxicity grading scale will be used for assessing AE severity; reported AEs that are not specifically listed in the WHO toxicity grading scale will be assessed using the AE severity grading scale shown in table 2.
Adverse event severity grading scale for events not specifically listed in the WHO toxicity grading scale
Statistical analyses
The primary endpoint of assessing the relationship of plasma emicizumab concentrations with CWA and TGA parameters will be evaluated in an exploratory manner. No confirmatory hypotheses have been established since the study is exploratory in nature. Continuous data will be presented using summary statistics including means, medians, ranges, SD and 95% CIs while categorical data will be presented using summary statistics such as frequencies and percentages. An interim analysis will be performed to evaluate coagulation potential in blood samples from participants who entered the maintenance phase of emicizumab treatment at the age of <12 months after enrolment in the study and will report on the safety of emicizumab up to the data cut-off point.
Ethics and dissemination
The HINODE study was approved by the MINS Clinical Trial Review Committee, a non-profit organisation (approval no. 230214) and will be conducted in compliance with the Declaration of Helsinki (Japan Medical Association translation), the Act on the Protection of Personal Information and the Guidance of Ethical Guidelines for Medical and Biological Research Involving Human Subjects. Written informed consent for participation in the study will be obtained from the legally acceptable representative after being provided with a clear explanation of the clinical study. The study protocol will be submitted for centralised review and approved by the central Institutional Review Board (IRB). Any changes to the protocol or informed consent form must be approved by the IRB. The results of the study will be summarised in a clinical study report that will be shared with the principal investigators and collaborating institutions. Results will be communicated through publication in scientific/medical journals and at international congresses.
Patient and public involvement
There was no patient or public involvement in the design and conduct of this study.
Registration
The HINODE study is registered in the Japan Registry of Clinical Trials (jRCT1031230264; date of registration: 29 July 2023).
Discussion
The HINODE study will generate data on coagulation capacity with emicizumab in infants just after birth whose haemostatic system is maturing during this stage of physiological development. Previous studies using CWA and TGA have shown the in vitro coagulation potential of emicizumab. A study using a neonate-HA model plasma incubated with emicizumab reported heterogenous CWA and TGA results30 while in another study using plasma from infants and toddlers with HA, CWA and TGA found improvements in coagulant potential following the addition of emicizumab.18 The HINODE study will use both CWA and TGA to measure coagulation potential in emicizumab-treated participants; CWA is a versatile and economical technique that is commonly used as it can be easily implemented in fully automated coagulation testing equipment in clinical laboratories.26 The study will also use anti-emicizumab idiotype antibodies to measure coagulation capacity without emicizumab interference, allowing accurate measurements of the original procoagulant and anticoagulant factor activity in participants.27 31
The real-world data from the HINODE study will build on the findings from previous clinical trials in paediatric participants with HA (aged ≤12 years)19 20 and from the HAVEN 7 study21 which enrolled newborns and infants up to the age of 12 months. While, in HAVEN 7, participants received a maintenance dose of emicizumab 3 mg/kg Q2W for the first 52 weeks, the HINODE study will provide clinical data on prophylactic emicizumab at all three approved maintenance doses (1.5 mg/kg QW, 3 mg/kg Q2W and 6 mg/kg Q4W). Additionally, the HINODE study will help to clarify the safety of emicizumab therapy in this age group and to support the importance of initiating treatment early to protect against life-threatening ICH and other bleeding after birth.
The timing of when to start emicizumab prophylaxis is a matter of debate among haemophilia-treating physicians. The World Federation of Hemophilia recognises that the subcutaneous route of administration of emicizumab may allow initiation of prophylaxis from birth and the National Bleeding Disorders Foundation’s Medical and Scientific Advisory Council supports that infants should be considered for emicizumab prophylaxis at any time after birth; however, both organisations acknowledge that further research on initiation of emicizumab in newborns is required.1 32 The present study will further evaluate the impact of low FIX concentration levels in the early life of infants on emicizumab using global coagulation assays. Additionally, although the early start of emicizumab prophylaxis during the first 3 months of life may help to reduce the risk of ICH, not all patients are diagnosed early and neonates are at the highest risk of ICH at the time of delivery before emicizumab prophylaxis can even be initiated.33 In a single-centre open-label prospective study including 27 infants under 1 year of age with severe HA, four cases of ICH occurred, all within the first 7 days following delivery and prior to emicizumab prophylaxis being initiated; however, no new cases of ICH were observed after starting emicizumab.24 The median age at prophylaxis initiation was 7 months among the 27 infants enrolled between 2018 and 2023, although this decreased to 3 months in 2023 suggesting a trend towards starting emicizumab at a younger age.24 A survey conducted among paediatric haemophilia treatment centres in the PedNet Registry including members across Europe, Canada and Israel demonstrated that most centres (85%) started emicizumab prophylaxis in patients before 1 year of age but only 30% started it before 6 months of age and only 10% before the age of 3 months.33 Therefore, further research is required into early treatment optimisation with the aim of reducing the incidence of bleeds during the first year of a child’s life which will have implications for long-term joint health and quality of life.
A strength of this study is that it will provide data from an estimated sample size of 50 patients up to 12 months of age with congenital HA making this one of the largest studies in newborns and infants treated with emicizumab. Additionally, the study will use multiple coagulation tests including CWA and TGA to obtain a broad picture of the coagulation capacity of infants receiving emicizumab during this time period. A limitation of the HINODE study is that since it is an observational study, it may result in a low accumulation of cases of <6 months of age. In addition, a sufficient volume of appropriate blood sampling is not easy to obtain in infants and the frequency of venous punctures needs to be minimised in infants with HA. Owing to difficulties in completing all planned testing for samples obtained at any time point, sequential blood sampling will be performed at only two time points: <6 months and 6 to <12 months of age.
In conclusion, the HINODE study will provide data on the coagulation potential of emicizumab in infants from birth to 12 months of age as well as on its safety which could allow for earlier initiation of prophylaxis, reducing the risk of life-threatening bleeds and delaying or minimising exposure to FVIII replacement, thus reducing the risk of developing FVIII inhibitors.
Ethics statements
Patient consent for publication
Acknowledgments
The authors would like to thank the study participants and their families as well as the study investigators, research coordinators and nurses. Third-party medical writing assistance was provided by Flaminia Fenoaltea, MSc and Katie Smith, PhD, of Ashfield MedComms, an Inizio company, and was funded by Chugai Pharmaceutical Co., Ltd.
References
Footnotes
X @Daisuke Nosaka
Contributors SO, MT, MI, HI, DN, KI, CM and KN contributed to study design. MT, MI, HI, DN, KI, CM and KN contributed to study conduct. MT, HI and KN contributed to recruitment and follow-up of patients. MT, HI and KN contributed to data collection. SO, MT and KN contributed to data analysis and interpretation. SO acted as guarantor. All authors have reviewed and approved the manuscript prior to submission and agree to be accountable for all aspects of the work.
Funding The study is funded by Chugai Pharmaceutical Co., Ltd.
Competing interests SO received a grant (scholarship donation), speaker fees and manuscript writing fees from Chugai Pharmaceutical Co., Ltd. MT received research grants and speaker fees from Chugai Pharmaceutical Co., Ltd. MI received speaker fees from Chugai Pharmaceutical Co., Ltd. DN, KI and CM are employees of Chugai Pharmaceutical Co., Ltd. KN received research grants from Chugai Pharmaceutical Co., Ltd; personal fees from F. Hoffmann-La Roche Ltd; grants, personal fees and non-financial support from Sysmex; grants and personal fees from Sekisui Medical; honoraria for lectures from Chugai Pharmaceutical Co., Ltd, Takeda, CSL Behring, Novo Nordisk A/S, Sanofi S.A., KM Biologics, Fujimoto Pharmaceutical, Bayer AG, Bioverativ, Shire; and is a co-inventor of a patent related to an anti-FIXa/FX bispecific antibody. All other authors declare no competing interests.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.